1. Introduction
N-nitrosamines are carcinogenic compounds that were detected in drugs for the first time in July 2018 [
1]. The International Agency for Research on Cancer (IARC) placed
N-nitrosodimethylamine (NDMA) and
N-nitrosodiethylamine (NDEA) in group 2A (possibly carcinogenic to humans), and the European Union have placed them in category 1B (substances allegedly carcinogenic), whereas the US Environmental Protection Agency (EPA) classified these two compounds into Category B2 (possibly carcinogenic to humans). The identification of these impurities in angiotensin receptor blockers (ARBs), mainly sartans with a tetrazole ring, and in metformin and ranitidine has led to the recall of certain batches of active pharmaceutical ingredients (APIs) from the market [
2]. Since then, steps have been taken to avoid the presence of
N-nitrosamine impurities in human medicines [
2]. Marketing authorisation holders (MAHs) should now review their manufacturing processes to evaluate the risk of
N-nitrosamine formation in APIs and finished products, perform confirmatory testing on the products identified to be at risk and, finally, apply for any necessary changes to the manufacturing process using the appropriate regulatory procedures. In February 2021, the European Pharmacopeia (Ph. Eur.) commission revised the five monographs on sartans with a tetrazole ring, namely Valsartan (2423), Losartan potassium (2232), Irbesartan (2465), Candesartan cilexetil (2573) and Olmesartan medoxomil (2600), using the rapid revision process. A reference to the general section “2.5.42. N-Nitrosamines in APIs” was introduced in the “Production” section to assist manufacturers. The five revised monographs became legally binding on 1 April 2021 [
3].
There are many identified sources of
N-nitrosamines in drug products [
4], such as the use of nitrites, nitrates or other nitrosating agents in the presence of secondary or tertiary amines or quaternary ammonium salts within the same or different process steps.
N-nitrosamines can also form as a result of the use of contaminated starting materials, intermediates and raw materials (e.g., solvents, reagents and catalysts) in the API manufacturing process or even by storing drug products in contaminated blister packaging materials [
4]. Recently, Zmysłowski et al. [
5] stated that excipients used in drug products are not supposed to be a direct source of
N-nitrosamines; however, they could possibly act as precursors of the reaction between dimethylamine (DMA) and nitrites/nitrates. According to the World Health Organization (WHO), the interim allowable intake limits for several
N-nitrosamines [
6] are 96.0 ng/day for NDMA and 26.5 ng/day for NDEA. Consequently, the acceptable
N-nitrosamine content in a drug product depends on its maximum daily dose, but the NDMA and NDEA levels should, in general, be as low as possible. To achieve this goal, comprehensive and sensitive analytical methods providing such low limits of quantification (LOQ) should be developed.
A few months after the so-called ‘sartan crisis’, a review article summarised the analytical methods developed so far to determination
N-nitrosamines in samples from various sources, such as drinking water, meat, beer, cigarette and tobacco smoke, rubber teats and soothers, cosmetic products, atmospheric particulates, soil and sewage sludge, as well as human urine [
7]. Due to the volatility of
N-nitrosamines, most of the reported methods utilised gas chromatography (GC) separation accompanied by different detectors, such as a mass spectrometer (MS), nitrogen-phosphorous detection (NPD) or a nitrogen chemiluminescence detector (NCD), which is also called a thermal energy analyser (TEA). The use of the latter is limited to only nitrogen compounds [
8], which results in its low popularity in control laboratories, such as Official Medicines Control Laboratories (OMCLs). There are also a few
N-nitrosamines detection methods using high-performance liquid chromatography (HPLC), which is widely used in OMCLs and by manufacturers of APIs and finished products.
N-nitrosamines can be monitored by a common UV-Vis detector at 230 nm, but it is difficult to obtain the required LOQ at the μg/g level without a preconcentration of the sample and baseline fluctuations, making it difficult to obtain reproducible results with acceptable RSD values for repeatability. MS, MS/MS and TEA detectors can also be used after coupling to HPLC systems. Although the European Directorate for the Quality of Medicines (EDQM) mentions GC-MS and LC-MS on the list of ad-hoc OMCL Network projects [
9], in practice, the mass detector is very expensive and, therefore, not easily accessible in every analytical laboratory. LC/MS also has limitations related to the optimal
m/
z values detected, which should be greater than 100 Da, while the mass of the NDMA molecule (74 Da) is relatively small and may cause technical problems with matrix interference and ion suppression effects. The alternative methods of detection utilise the derivatisation and monitoring of luminescence of the resultant compounds. In one case,
N-nitrosamines are deprived of the NO group by means of hydrobromic acid (HBr) in acetic acid, and the liberated amines are then reacted with a fluorescent compound (pre-column derivatisation) to form derivatives that are finally separated in a chromatography column and monitored by a fluorescence detector. In an alternative method,
N-nitrosamines are first separated on an HPLC column, then denitronised with a UV lamp and, finally, derivatised and observed by a chemiluminescence detector (post-column derivatisation).
N-nitrosamines can also be determined using capillary electrophoresis [
10] and non-separational techniques, but the latter only enable us to determine the sum of
N-nitrosamines rather than a concentration of each individual one [
7].
This work focuses on the development and validation of a method for the determination of
N-nitrosamines by means of pre-column derivatisation with fluorescence detection. The use of spectrofluorimetric detectors is widespread in OMCLs and manufacturers of APIs and finished products, because they are commonly used in the determination of compounds with a fluorophore, such as aromatic compounds and their derivatives. Due to the very good detectability and specificity of determinations with a fluorescence detector, non-fluorescing compounds are converted into their fluorescent derivatives. There are many derivatisation agents that react with the amine group liberated from
N-nitrosamines by cleavage with HBr in acetic acid, including 5-(dimethylamino)naphthalene-1-sulfonyl chloride (dansyl chloride) [
11,
12,
13], 2-(11H-benzo[a]carbazol-11-yl) ethyl carbonochloride or 1-fluoro-2,4-dinitrobenzene [
7]. Fluorenylmethoxycarbonyl chloride (Fmoc-Cl) has been widely used since 1972 in the determination of short-chain aliphatic amines, biogenic amines, catecholamines [
14] and amino acids [
15]. Due to its reaction with amine groups, it can be used in the determination of particular classes of drugs, such as aminoglycosides, amphetamines, sulfamethazine, macrolide antibiotics, bisphosphonates [
14] and sodium alendronate (Ph.Eur. 01/2017: 1564) [
16]. To the best of our knowledge, there are no reports on the determination of
N-nitrosamines using Fmoc-Cl. However, there are a few reports on the determination of DMA, which is a product of the denitrosation of NDMA [
17,
18,
19,
20]. The aim of our research is to develop a reproducible analytical method that would provide an appropriate LOQ, as required by the European Medicines Agency (EMA), and would not require expensive and sophisticated equipment, such as GC-MS and LC-MS. For this purpose, the most popular reagent for the derivatisation of
N-nitrosamines, dansyl chloride and the first used for this purpose, Fmoc-Cl, were selected. The novelty of this work relies on the combination of the aforementioned denitrosation process and derivatisation with Fmoc-Cl to create a new method of
N-nitrosamine determination in enalapril maleate.
There are many active substances that can become a potential source of
N-nitrosamines contamination, many of which are amine-based pharmaceuticals [
7]. For this reason, we have drawn our attention to enalapril maleate, which contains both secondary and tertiary amine groups (see
Figure 1). Enalapril maleate belongs to the class of angiotensin-converting enzyme (ACE) inhibitors and is used in the treatment of hypertension, diabetic kidney disease and heart failure [
21]. In addition, after the sartan crisis, according to the guidelines, some patients switched from contaminated ARBs to noncontaminated ARBs or other antihypertensives, such as ACE inhibitors (including enalapril) or calcium channel blockers. Since enalapril maleate is a blockbuster with antihypertensive drugs and is prescribed to be taken daily for a long time [
22], patients may be at risk of a chronic intake of carcinogenic impurities; hence, it is extremely important to carefully monitor the
N-nitrosamines level in this API. The development of a reproducible method that could be widely used by OMCL gives patients the chance to receive a safe treatment alternative. The upper limit of the NDMA content for enalapril maleate can be obtained by dividing the maximum allowable intake limit of NDMA (96 ng/day) by the maximum daily dose for enalapril (40 mg/day), which gives an acceptable level of 2.4 μg/g.
2. Materials and Methods
2.1. Materials
Enalapril maleate (99.7%) was purchased from Zhejiang Huahai Pharmaceutical (Linhai, China). NDMA (200 µg/mL in methanol), NDEA (5000 µg/mL in methanol), dimethylamine (DMA, 40% wt. % in H2O), diethylamine (DEA, ≥99.5%), Fmoc-Cl (≥99.0%, BioReagent), sodium hydroxide (NaOH, ≥98%) and sodium acetate (CH3COONa × 3 H2O, ≥99.0%) were obtained from Sigma-Aldrich (Saint Louis, MO, USA). Acetonitrile (ACN, HPLC gradient grade, ≥99.9%), dichloromethane (DCM, HPLC, ≥99.8%), hydrobromic acid (HBr, ≥ 48%), sodium bicarbonate (NaHCO3, ≥99.7%) and sodium sulphate (Na2SO4, ≥99.0%) were obtained from Honeywell (Charlotte, NC, USA). Glacial acetic acid (CH3COOH, 99.9%) was purchased from AppliChem (Darmstadt, Germany), dansyl chloride was obtained from both Acros Organics (98%, Geel, Belgium) and Sigma-Aldrich (BioReagent, ≥99%) and boric acid (H3BO3, reagent grade) was purchased from Merck (Darmstadt, Germany), while acetone (analytic grade) was obtained from STANLAB (Lublin, Poland). Deionised water was obtained from a Labconco System by Millipore (Bedford, MA, USA) and was boiled before being used to remove possible contaminations with volatile DMA and DEA.
2.2. Equipment
The HPLC analysis was performed using a Shimadzu Nexera-i LC-2040C 3D (Japan) liquid chromatograph with PDA and an external Shimadzu RF-20A fluorescence detector.
All glassware used in the experiment was carefully washed before the analysis in two steps—sonication for 30 min in deionised water at 80 °C and sonication in DCM for 15 min—to get rid of the residual N-nitrosamines or secondary amines from the laboratory glassware used for testing.
2.3. Nitrosamine Extraction from APIs
First of all, 250 mg of enalapril maleate was suspended in 5 mL of DCM and vortexed for 1 min to give a white homogeneous suspension at a concentration of 50 mg/mL. Since enalapril is practically insoluble in DCM (<0.1 mg/mL) [
16], the suspension was filtered through a paper filter into a 5-mL volumetric flask. DCM was then added to the mark. The resulting clear solution was completely transferred to a separation funnel, and 5 mL of acetate buffer, pH 5.6 was added. The 0.1-M acetate buffer, pH 5.6 was prepared freshly before use by dissolving sodium acetate trihydrate in water and adding acetic acid to obtain the desired pH value. The mixture in the separation funnel was shaken for 5 min. After the extraction process, the separation funnel was left for 30 min to improve the phase separation, the lower phase (DCM) was then collected and 1-mL samples (
n = 3) were taken from each separating funnel.
2.4. Preparation of Standard Solutions
Standard solutions of
N-nitrosamines were prepared in amber flasks by dissolving NDMA and NDEA in DCM and subjecting them to the same process of extraction as the sample solutions (
Section 2.3). There are two ways to express their concentrations, in a solution in ng/mL and in relation to API powder expressed in μg/g. The International Union of Pure and Applied Chemistry (IUPAC) recommends avoiding the use of ppm or ppb and using SI units instead, such as μg/g or μg/mL. This approach is particularly useful in this work, as it allows avoiding confusion and directly indicates whether the concentration is related to the solution or to the API powder.
A blank solution was prepared by shaking 5 mL of DCM with 5 mL of 0.1-M acetate buffer, pH 5.6 and taking three 1-mL samples from the DCM phase.
2.5. Denitrosation of NDMA and NDEA
The denitrosation reagent was prepared by dissolving 1 mL of HBr in 10 mL of CH
3COOH and stored in an amber flask in a refrigerator. Then, 1 mL of standard/sample solution in DCM was transferred to a 1.5-mL amber HPLC vial, and 50 µL of denitrosation reagent was added. The vials were then capped with a filled screw cap without any holes for an HPLC needle and without a rubber septa (which could potentially be contaminated with amines or
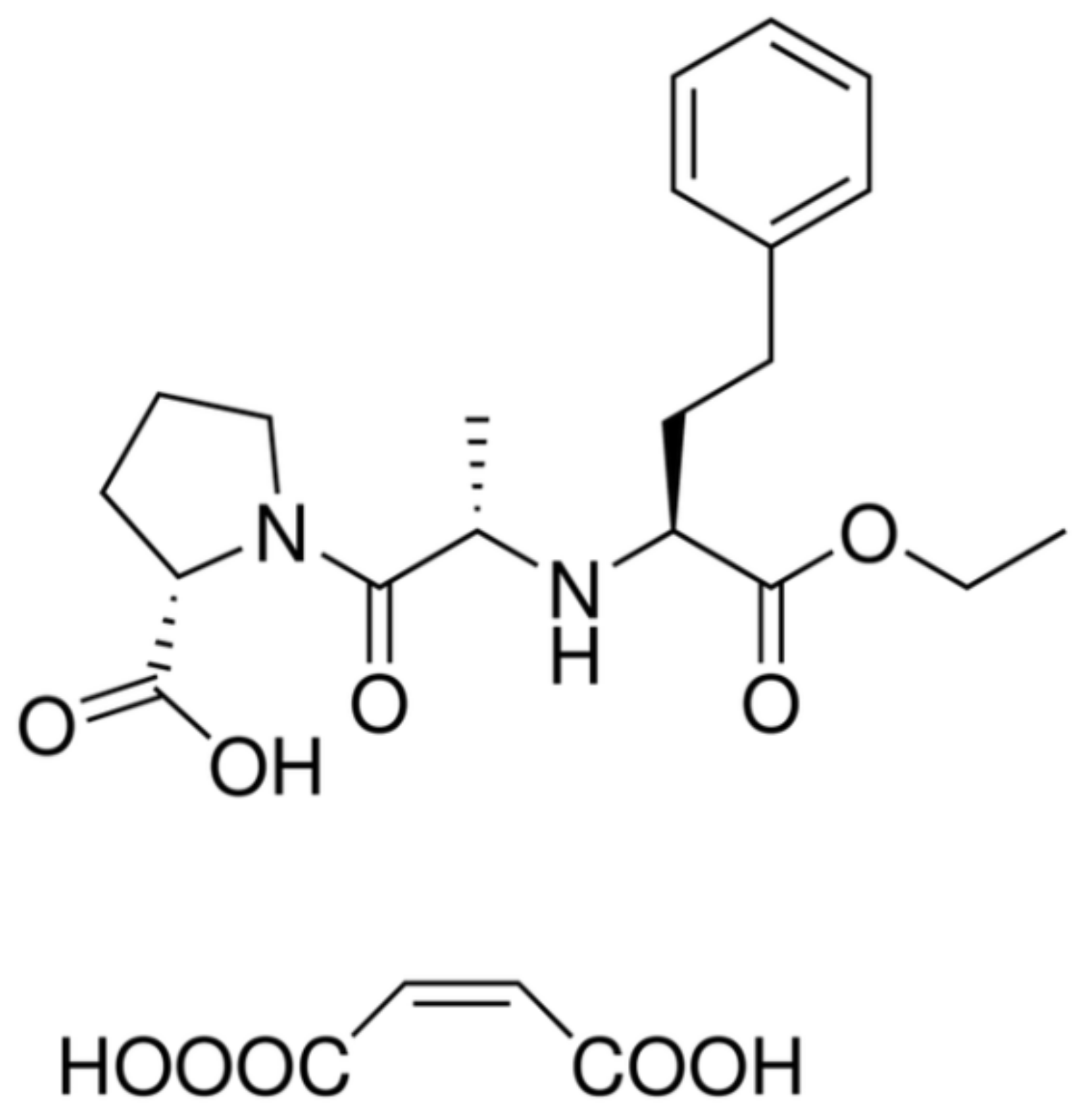
N-nitrosamines) and shaken using a vortex for 10 s. The vials were left capped for 30 min at room temperature in the dark and then heated for 20 min at 60 °C. Afterwards, the vials were opened and left, usually for more than 180 min at 60 °C in an oven, to completely evaporate the solvent and the acids. The processes of denitrosation and derivatisation are illustrated in
Figure 2.
2.6. Derivatisation Process with Dansyl Chloride
After preparation of the standard, denitrosation and cooling the vial to room temperature, 50 µL of 1-M NaOH and 200 µL of 0.5-M NaHCO3 were added to obtain the desired pH to achieve the appropriate conditions for derivatisation. Then, 150 µL of 0.5-mg/mL dansyl chloride in acetone was added. The solution was heated for 30 min at 40 °C in an oven and then placed in the HPLC autosampler. The total volume of the sample was 0.4 mL; thus (given that the initial volume was 1 mL), the sample was concentrated during the previous two steps by a factor of 2.5.
2.7. Derivatisation Process with Fmoc-Cl
A 0.2-M borate buffer, pH 8.5 was prepared freshly before use by dissolving boric acid in water and adding 10-M NaOH. Then, 200 µL of 0.2-M borate buffer, pH 8.5 and 200 µL of 5-mM Fmoc-Cl solution in ACN were added to the vial with a sample after evaporation. ACN was used, as it is regarded as a better solvent for Fmoc-Cl than acetone [
15]. The solution was heated for 60 min at 40 °C in an oven and then placed in the HPLC autosampler.
2.8. Optimisation of Chromatographic Conditions for Both Classes of Derivatives
The chromatographic conditions are summarised in
Table 1. The column, mobile phase and excitation/emission wavelengths were taken from the literature [
12,
15]. The gradient program, injection volumes and sensitivity of the fluorescence detector were the subject of optimisation and will be discussed in
Section 3.
2.9. Method Validation
The linearity, repeatability and LOQ of the method were determined by preparing a calibration curve for the standard solutions with concentrations ranging from 1 to 120 ng/mL corresponding to 0.02–2.4 μg/g in the API. The 2.4-μg/g value is the maximum allowable limit of NDMA in enalapril. The standards were subjected to extraction, denitrosation and derivatisation with Fmoc-Cl. Two independent extractions were performed for each concentration, and three samples were taken from each separation funnel.
The accuracy of the method was performed as follows: 250 mg of enalapril maleate was weighed into a 5-mL volumetric flask, and a certain amount of
N-nitrosamines stock solution (10 μg/mL) was added to the bottom of the flask using a syringe with a metal needle, until the final
N-nitrosamines concentrations were equal to 0.5 μg/g, 1 μg/g and 2.4 μg/g with respect to enalapril maleate. The flasks were stoppered, shaken by hand and left for 30 min to allow the
N-nitrosamines to penetrate the entire volume of the powder. The flask was then filled to the mark with DCM, and the Fmoc-Cl sample preparation procedure was followed as described in
Section 2.3,
Section 2.5 and
Section 2.7. Each single concentration experiment was performed in duplicate, and three samples were taken from each separation funnel. The peak areas of the standard solutions from the linearity study, at concentrations equal to those applied in this experiment, were used as a reference. The recovery factors were calculated based on the peak areas obtained with the spiked samples and standard solutions.
3. Results and Discussion
This section presents not only the obtained results, but also discusses several critical points affecting the repeatability of the method, which are not covered in similar articles.
3.1. Extraction of NDMA and NDEA
The extraction step seems necessary to remove the secondary amines, which may be residual impurities in some APIs and may interfere with the corresponding N-nitrosamines, as they can also be derivatised to form the same derivative as the corresponding N-nitrosamines.
The effectiveness of removing the secondary amines from the assayed samples was proven by extracting the solution of amines (DMA and DEA) at a concentration of 20 μg/g in DCM with the acetate buffer, pH 5.6. This pH value was chosen on the basis of the pKa values for N-nitrosamines (pKa 3.52) and amines (pKa 10.52), which provides intermediate conditions in which N-nitrosamines should be neutral (and more likely to be found in the DCM layer), while amines should be charged (and more willing to be dissolved in an aqueous layer). Indeed, when the DCM layer from the separation funnel was subject to denitrosation and derivatisation, its chromatogram was indistinguishable from that of the blank, indicating that the secondary amines went into the aqueous phase and did not interfere with the N-nitrosamines. Special care must be taken during the process of extraction in order to avoid the formation of an opaque emulsion. The residual water in the DCM layer led to erroneously increased and unrepeatable areas of the analysed peaks, and the addition of Na2SO4 to remove residual water from the organic layer surprisingly had the same negative effect. Only clear solutions after extraction allow consistent results to be obtained in the tested samples.
3.2. Denitrosation of NDMA and NDEA
The denitrosation process (see
Figure 2) was carried out as previously reported [
12,
13] but with different amounts of reagents and a longer evaporation time. Until now, this method has been used in water research, and it is now applied to determine the
N-nitrosamines in API.
It has been found that heating in the air causes residual acids (CH3COOH and HBr) to evaporate faster than in a water bath. It is absolutely necessary to allow the vials to dry completely; otherwise, the acids remaining at the bottom of the vial will lead to incorrect pH values in the next step and unrepeatable peak areas in the chromatograms.
Comparing the boiling points of the mixture components (hydrobromic acid (122 °C), CH3COOH (118 °C), DEA (55.5 °C), DCM (40 °C) and DMA (7 °C)), one can ask how acids can be evaporated without loss of the secondary amines. It was assumed that the amines liberated from the N-nitrosamines react with HBr to form hydrobromides, which are solids at 60 °C and do not evaporate from the vial but precipitate at the bottom.
3.3. Derivatisation Process with Dansyl Chloride (5-(Dimethylamino)Naphthalene-1-Sulfonyl Chloride)
In this work, two different methods of derivatisation were performed and compared. The first utilises dansyl chloride (see
Figure 2) and is based on the available literature on water analyses [
12,
13].
According to the literature, the pH does not influence the derivatisation process in the range of 9–13 [
12]. Using indicator paper, it was proven that the pH of the solution was between 10 and 11 after the addition of 0.1-M NaOH, 0.5-M NaHCO
3 and dansyl chloride.
In order to choose an appropriate amount of dansyl chloride in the vial, various volumes of dansyl chloride solution were added to the vial, and the effect was then analysed. In every case, 200 µL of 4-μg/g aqueous solution of DMA and DEA (standard) or 200 µL of boiled water (blank) were transferred to a HPLC vial. The appropriate amounts of 0.1-M NaOH and 0.5-M NaHCO
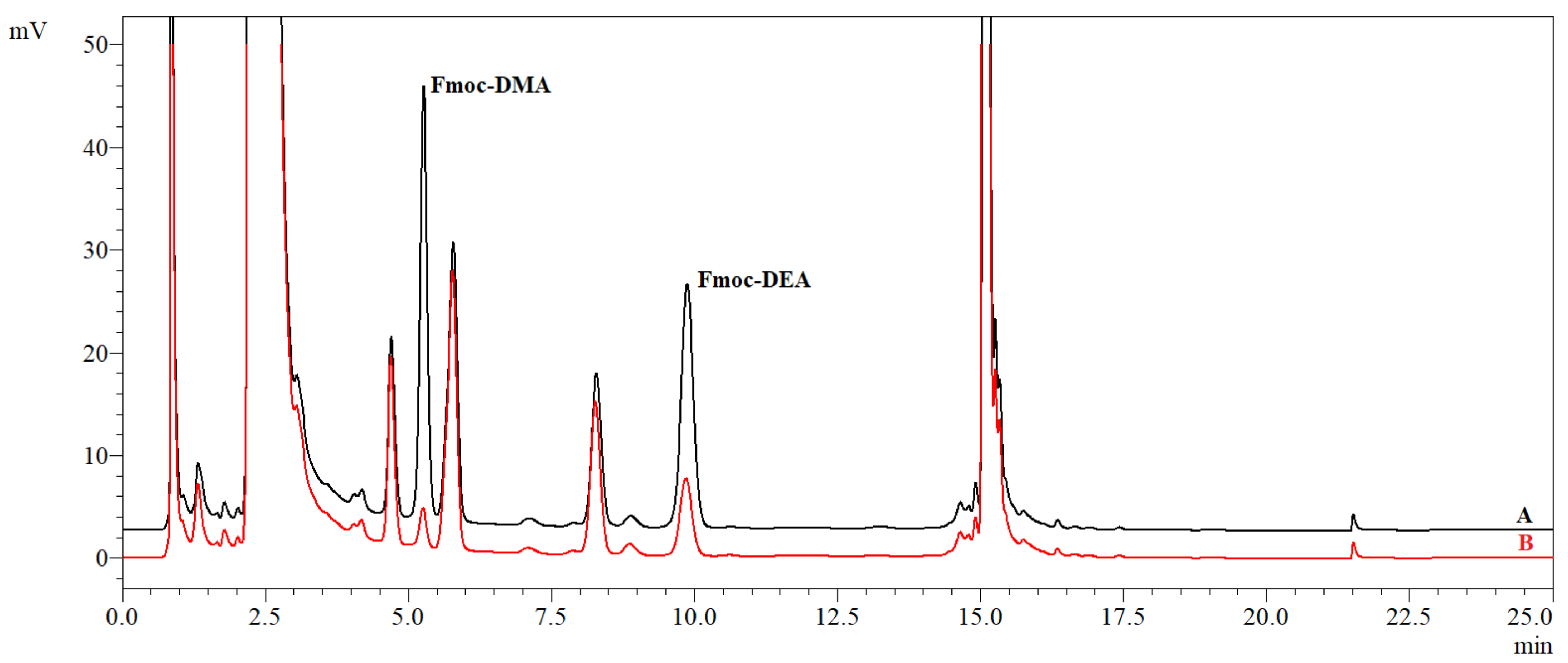
3 were then added to ensure the correct pH value, and an appropriate volume of acetone was added to make the same total volume for all the samples. The lowest concentration of dansyl chloride in the vial in this experiment was 5 μg/mL. With a decreasing amount of dansyl chloride used in the derivatisation process, the peak in the blank decreased more quickly than the peak in the standard (the ratio of these two is more beneficial and enables us to determine the lower amounts of NDMA). However, the reduction of the dansyl chloride concentration decreased the sensitivity of the method, especially for NDEA. This fact makes it difficult to determine the NDMA and NDEA at the same concentration level (see
Figure 3). On the other hand, NDEA is recognised to be more toxic than NDMA, allowing a lower daily intake limit for this
N-nitrosamine (26.5 ng/day vs. 96 ng/day), so it is more important to obtain a sufficient sensitivity for NDEA. Therefore, decreasing the concentration of dansyl chloride is not beneficial.
The influence of the derivatisation time on the peak areas was also checked, using standard solutions of NDMA and NDEA at concentrations ranging from 0 (blank) to 1 μg/g. The samples were denitronised, according to the procedure described above, and then, the solutions for derivatisation were added. Afterwards, the vials were capped with a standard cap for a screw top HPLC vial (with a hole for a needle and with a septa); vortexed for 10 s and heated for 30, 60, 90 or 120 min, respectively. Finally, the vials were cooled at room temperature, placed in the HPLC autosampler and analysed. The results are difficult to interpret and unrepeatable, however, as there is no trend relating to the change of the peak area and the time of heating, and the calibration curves plotted for the different periods of heating were nonlinear.
Chromatograms of the standard solution and blank sample after derivatisation with dansyl chloride are presented in
Figure 3. The mobile phase gradient begins with an isocratic step of 45% ACN, which was introduced in order to avoid problems with an ascending baseline under UV-VIS detection and to ensure a sufficient resolution between the neighbouring peaks. A lower ACN content would result in a longer analysis time and a decrease in the sensitivity of the method due to the broadening of the peaks for compounds with longer retention times. After the elution of the DEA derivative, the ACN concentration rises abruptly to 100% so as to elute any remaining impurities from the column. Finally, the column is stabilised for 5 min with the initial concentration of the mobile phase.
The use of dansyl chloride as a derivatising reagent has several drawbacks, which make the determination of amines/
N-nitrosamines at low concentrations difficult. Firstly, a blank sample (which is pure DCM without amines/
N-nitrosamines subjected to the same denitrosation and derivatisation processes as the standards and samples) shows a large and irreproducible peak at the same retention time as the DMA derivative. Since it could not be reduced by boiling the deionised water used to prepare the solutions each day, it was assumed that the dansyl chloride used was contaminated with DMA [
23], because the dansyl chloride at the C5 position has a dimethylamino substituent. Secondly, the responses of the dansyl derivatives were low, so it was useful to use the maximum sensitivity of the fluorescence detector. Finally, the derivatisation kinetic studies indicated a lack of linearity and reproducibility in the concentrations range of DMA and DEA below 0.2 μg/g (data not shown). Derivatisation with dansyl chloride proved to be an impractical method for the determination of
N-nitrosamines at the μg/g level without a preconcentration of the samples by means of solid-phase extraction. Although this method was mentioned in a review article summarising the NDMA determination methods [
7], it suffers from many technical problems, such as low sensitivity, the content of DMA derivatives in the blank solution and the disproportionate response of NDMA and NDEA. Therefore, in this work, studies with dansyl chloride were not continued.
3.4. Derivatisation Process with Fmoc-Cl
The analytical method of derivatisation with Fmoc-Cl was developed based on several reports concerning secondary amines [
17,
18,
19] and a review article [
15]. The novelty of this work relies on the combination of the denitrosation process mentioned earlier with the derivatisation with Fmoc-Cl to create a new method of
N-nitrosamine determination.
During the method development, it became evident that it is essential to use Fmoc-Cl of the highest available purity, based on our previous experience with dansyl chloride. When Fmoc-Cl 97% was used, a much greater peak from the DEA derivative was observed in the blank solution. Additionally, deionised water was boiled before each use to remove possible contaminations with volatile DMA and DEA.
Contrary to the rather weak response of dansyl chloride, Fmoc-Cl requires much lower values of sensitivity and gains in fluorescence detection. What is more, DMA and DEA can also be determined using a common UV-VIS detector (265 nm,
Figure 4); however, a fluorimetric detector offers better sensitivity (
Figure 5).
Three experiments were performed to determine the robustness of the derivatisation process. In the first stage, (i) a borax buffer (pH 9.3) and several borate buffers with the following pH values were prepared: 8.0, 8.5, 9.0 and 9.5. It was proven that the pH of the buffer did not significantly affect the of N-nitrosamines area in the standard solution (4 μg/g) or in the blank solutions. The same is true for the Fmoc-Cl concentration (ii) (which was tested using 0.2-mM, 1.0-mM, 5.0-mM and 10.0-mM solutions in ACN). The derivatisation carried out at room temperature gave a large peak in the chromatogram with a retention time of 10 min, which decreased with time (and disappeared completely after about 10 h). However, when the reaction took place at 40 °C and for at least 1 h, no peak appeared at all. Finally, (iii) the derivatisation kinetics studies showed that the influence of the test sample heating time in the 0.5–2-h range (30, 60, 90 and 120 min) is negligible both on the peak areas and linearity.
The gradient starts with an isocratic 55% ACN step to avoid the rising baseline problems under UV-VIS detection, provide sufficient resolution between the neighbouring peaks and achieve capacity factors in the range k’ ≈ 5–10 for Fmoc-DMA and Fmoc-DEA, respectively. The 100% ACN step is mandatory, because the very nonpolar impurities that arise during the derivatisation process must be eluted from the column. This is especially true for those compounds that would have a retention time of 25 min in an isocratic mode with 60% ACN.
Several different chromatographic columns, such as ACE C18-PFP, Synergi Polar RP and Luna Phenyl-Hexyl, were tested in the optimisation stage of the method but offered significantly poorer resolution using the same gradient program. On the other hand, decreasing the ACN content to improve the resolution resulted in a much longer analysis time or the necessity to apply a higher flow rate, which, in turn, resulted in a higher pressure unacceptable for older HPLC systems.
Fmoc-Cl proved to be a much better derivatisation agent, and the method was therefore fully optimised and validated only with the use of Fmoc-Cl. Interfering peaks in the blank solution were still present, but the intensity for these peaks was significantly lower than with dansyl chloride. These peaks may have come from impurities of the reagents used in this study; therefore, their areas could not be lowered.
3.5. Method Validation
3.5.1. Specificity of the Method
The specificity of the method was confirmed by overlaying chromatograms of the blank solution and a sample of enalapril maleate API (
Figure 6). There are more peaks in the sample chromatogram, but the peaks of interest (DMA and DEA Fmoc derivatives) have similar areas in both the blank and API samples (not fortified with
N-nitrosamines) and are well-separated from other impurities. This means that the
N-nitrosamines content determined in the enalapril maleate sample is below the LOQ level (0.05 μg/g). The reason for the additional peaks in the sample’s chromatogram is that enalapril and its impurities are not completely separated by DCM during filtration (enalapril maleate has very low solubility in this solvent, while
N-nitrosamines are highly soluble). In the next step, API (enalapril maleate) is subject to a reaction with HBr, and finally, enalapril with its degradation products containing primary and secondary amine groups, is derivatised to form fluorescent products. This is one of the critical points of the method, because, depending on the source of API or its synthesis process and its degradation products generated in the denitrosation process, the degradation products containing primary and secondary amines will be derivatised with Fmoc-Cl in exactly the same process as the resulting amines liberated from
N-nitrosamines.
3.5.2. Linearity, Precision and Sensitivity of the Method
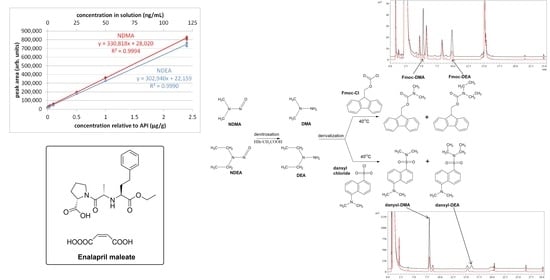
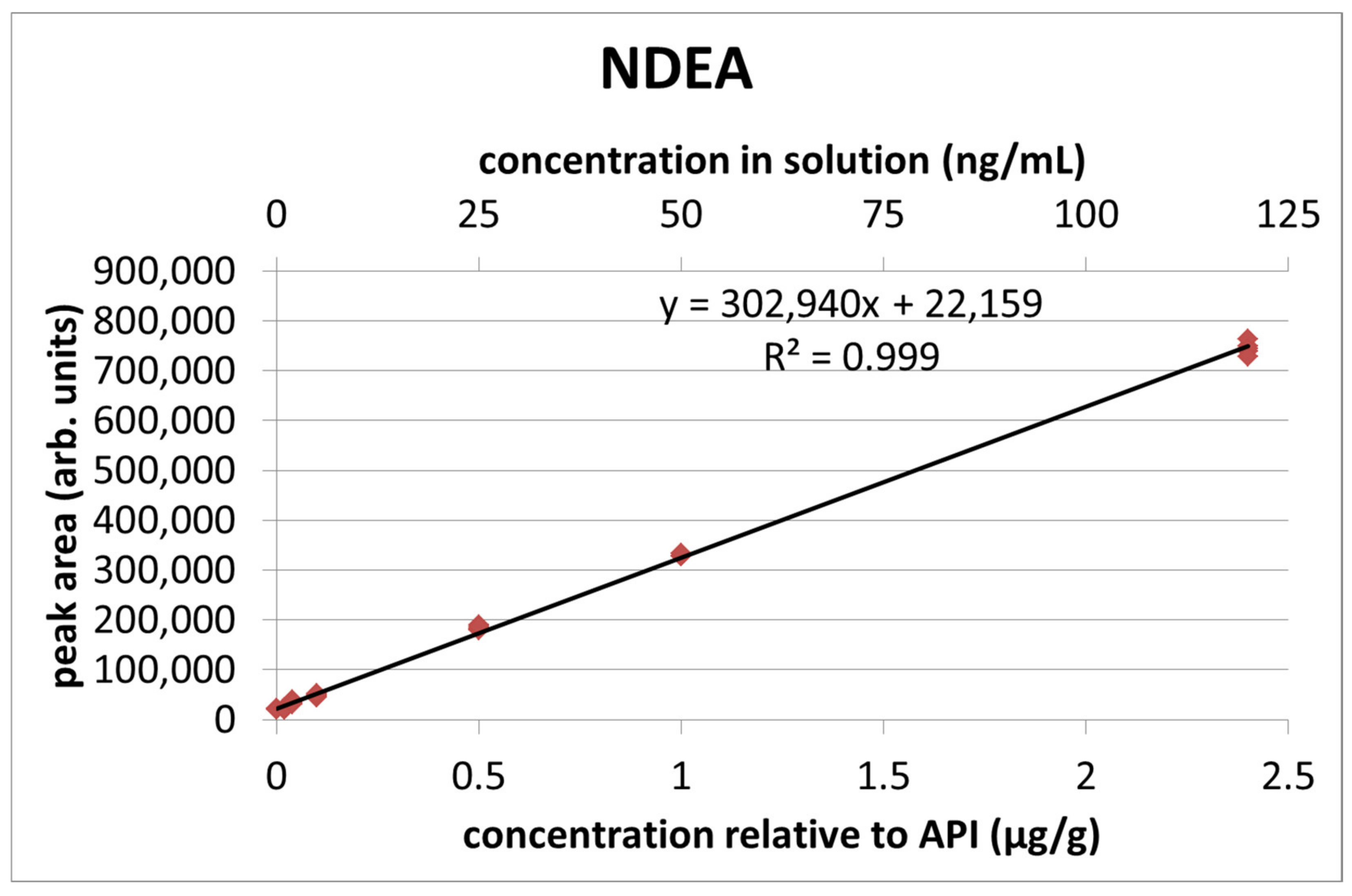
The calibration curves for NDMA and NDEA are presented in
Figure 7 and
Figure 8, while the numerical values are presented in
Table 2. Four samples had peak areas outlying from the others of the same concentration and were therefore discarded after using the Dixon test with a confidence level of 90%.
Table 2 presents two approaches to determine the method precision using the relative standard deviation (
). The first column presents deviations between the samples taken from the same separation funnel. The second column is calculated using peak areas of all samples of the same concentration and shows additional differences between results obtained from two separation funnels. Generally, the lower the concentration of
N-nitrosamines, the lower the precision. The highest
s were obtained for NDMA of a concentration of 0.02 μg/g (9.8%) and NDEA of a concentration of 0.04 μg/g (8.5%).
In the calibration curve, each individual sample was treated as one measuring point. The R
2 values were greater or equal to 0.999 for both nitrosamines (0.9994 for NDMA and 0.9990 for NDEA). The equations for the calibration lines are
y = 330,818
x + 28,020 with a residual standard deviation of 6375 for NDMA and
y = 302,940
x + 22,159 with a residual standard deviation of 7621 for NDEA (
x—concentration expressed in μg/g). The limit of quantitation was determined based on the ICH guidelines [
24] according to the equation:
where: σ—standard deviation of the intercept of the regression line (calculated by means of the REGLINP function in MS Excel) and
—slope of the calibration curve. The results were 0.038 μg/g for NDMA and 0.050 μg/g for NDEA.
The limit of detection was determined based on the ICH guidelines [
24] according to the equation:
The results were 0.013 μg/g for NDMA and 0.017 μg/g for NDEA.
3.5.3. Accuracy of the Method
The accuracy of the method was determined by calculating the recoveries by dividing the average peak area of the
N-nitrosamine in a spiked sample by a corresponding average area in a standard from the linearity study. The
of the recovery was calculated as follows:
Recovery is defined as the ratio of the observed mean test result to the true value. The recovery rates strongly depend on the sample preparation technique and sample concentration. The recoveries were in the range of 74.2 ± 4.2% to 101.6 ± 16.1% for NDMA and 90.6 ± 2.9% to 125.4 ± 7.4% for NDEA. The lowest recovery value was obtained for the highest concentrations of
N-nitrosamine (2.4 μg/g of NDMA), and the highest recovery was obtained for the lowest concentration (0.1 μg/g of NDEA).
Table 3 and
Table 4 present the raw experimental data and final results. All the recovery values are within the acceptance limit of 70–130% proposed in a recent article on the determination of
N-nitrosamines at a similar sub-μg/g concentration level [
5].
4. Conclusions
Derivatisation was performed with Fmoc-Cl, because it requires much lower values of the sensitivity and enhancement of fluorescence detection than dansyl chloride, and above all, reproducible results and corresponding sensitivity values of the method were obtained.
The specificity of the method was confirmed. The efficiency of removal of the secondary amines was proven by performing the extraction in DCM with acetate buffer, pH 5.6.
The robustness of the derivatisation process was investigated with respect to various borate buffers with a pH from 8.0 to 9.5 and borax solution (pH 9.3), as well as various concentrations of Fmoc-Cl, which were tested in the range of 0.2 mM–10.0 mM in ACN, and the effect of a heating time ranging from 0.5 to 2 h on the peak areas and linearity. The derivatisation process is insensitive to small variations in the pH, Fmoc-Cl concentration and heating time.
A satisfactory method linearity was obtained, where the R2 values were greater or equal to 0.999 for both nitrosamines (0.9994 for NDMA and 0.9990 for NDEA), with a LOQ level at 0.038 μg/g for NDMA and 0.050 μg/g for NDEA. The precision decreased with the concentration to a maximum level of about 10%. The recoveries were in the range of 74.2 ± 4.2% to 101.6 ± 16.1% for NDMA and 90.6 ± 2.9% to 125.4 ± 7.4% for NDEA.
Several critical points affecting the reproducibility of the method were discovered that have not been included in similar papers. The appropriate preparation of the glassware, gentle shaking of the separation funnel, full evaporation of the denitrosating agent and use of Fmoc-Cl of the highest available purity were found to be crucial in order to achieve a satisfactory method performance at the sub-μg/g level.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}