
Abstract
The epithelial–mesenchymal transition (EMT) is a molecular process connected to higher expression of vimentin and increased activity of transcription factors (Snail, Twist) which restrains E-cadherin. EMT has been linked to prostate cancer metastatic potential, therapy resistance, and poor outcomes. Kinetin riboside (9-(b-dribofuranosyl)-6-furfurylaminopurine, KR) is a naturally occurring cytokinin, which induces apoptosis and shows strong antiproliferative activity against various human cancer cell lines. To establish the effect of KR on human prostate cell lines, expression of, e.g. AR, E-, N-cadherins, Vimentin, Snail, Twist, and MMPs, was analysed at mRNA and protein levels using Western Blot and RT-PCR and/or RQ-PCR techniques. KR inhibited the growth of human prostate cancer cells, but also, to a small extent, of normal cells. This effect depended on the type of the cells and their androgen sensitivity. KR also decreased the level of p-Akt, which takes part in androgen signalling modulation. The antiapoptotic Bcl-2 protein was down-regulated in cancer cell lines, while that of Bax is up-regulated upon KR exposure. KR contributed to re-expression of the E-cadherin as well as to significant changes in cell migration. Taken together, our results indicate for the first time that KR can be proposed as a factor for signalling pathways regulation that participates in the inhibition of development of aggressive forms of prostate cancer, and may alter the approach to therapeutic interventions. We propose KR as a potent inhibitor of EMT in human prostate cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to the data provided by National Cancer Institute [1] in 2019, prostate cancer (PC) stands for 9.9% of all new cancer cases and is responsible for as many as 5.2% of all cancer deaths. Currently, one of the most commonly employed treatments for high-risk localised and locally advanced PC is androgen deprivation therapy (ADT) [2]. It is based on great dependence of PC development on androgen influence [3]. Although initial PC response to ADT may involve 80% cases, its duration usually lasts beyond 12 to 18 months [4], implying the need of introducing second line therapy, as well as a significantly worsening prognosis. Failure of ADT defines a castration-resistant prostate cancer (CRPC), a tumour that grows independently of exogenous androgens. At molecular level, this transformation may involve different patterns such as androgen receptor (AR) gene mutation or amplification, changes in expression of AR coregulators, increased expression of steroidogenic enzymes, increase in signalling through pathways that activate AR independently of androgens, as well as augmented signalling that bypasses AR [5]. Apart from acquiring resistance to ADT, androgen independence (AI) development also grants an independent survival advantage for PC. Interestingly, AR may also facilitate negative impact during PC progression [6], and thus a release from AR dependence; the following loss of the receptor expression may actually be beneficial for cancer. AI was described to develop in androgen-sensitive LNCaP prostate cancer cell line upon androgen deprivation, which was accompanied with enhanced proliferation rate, invasiveness, metastasis formation, and induction of epithelial–mesenchymal transition [7].
Alongside AI, epithelio-mesenchymal transition (EMT) was described as a vital part of PC development. It was linked to PC invasiveness, distant metastasis formation, chemoresistance, and poor clinical outcomes, proving its importance in disease progression [8]. Moreover, although EMT incidence in PC cells is sometimes attributed to androgen influence, it was also shown to be closely related to and enhanced by androgen independence development [8]. EMT was primarily described as a physiological process inherent to cellular differentiation during embryogenesis [8]. However, it is also involved in tissue morphogenesis, wound healing, and in pathological possesses such as fibrosis and cancer progression. It is also supposed to take part in many different diseases [9]. EMT is basically characterised by loss of epithelial cellular traits with subsequent gain of mesenchymal ones. These include loss of cellular polarity and adhesion, cytoskeleton and cellular junctions reorganisation, as well as increased cellular motility [9]. A lot of research was conducted to track molecular changes associated with EMT and those that govern this process. Among them, activation of transcription factors (TFs) like Snail, Slug, Twist, and Zeb2 seems to play a central role [9]. It seems that components of many molecular pathways including TGFβ, integrins, c-Src, Fak, PI3K-Akt, Rac, cdc42, MAPKs, mTOR, RTKs, GSK3β, β-catenin, Notch, Hedgehog, HIF1α, NfχB, as well as reactive oxygen species (ROS) may converge at these four TFs to regulate EMT [10]. Upon activation by either signalling pathway or epigenetic changes, Snail, Slug, Twist, and Zeb1 mediate changes in gene expression allowing epithelial cells to gain mesenchymal traits. Some of such changes governed by these TFs are up-regulation of vimentin, matrix metalloproteinases (MMPs) and N-cadherin (N-cad) with subsequent down-regulation of E-cadherin, (E-cad), cytokeratins, claudins and occludins [10]. In case of cancer cells, these together promote invasiveness, metastatic potential, treatment resistance, and probably cancer stem cell phenotype, and may be implicated in tumour dormancy [9]. Furthermore, EMT may play a vital role in the biology of circulating tumour cells (CTCs)—a subpopulation of cancer cells able to survive in patient blood and possibly to spread to distant organs to form metastases [9]. Importantly, EMT in cancer is a reversible process and undergoing mesenchymal–epithelial transition (MET) may be required for CTCs to form metastases [9]. It is also a matter of concern what the prime signal to induce EMT is as both extracellular (f.i. TGFβ, MMPs, ROS) and intracellular cues take part in EMT amplifying one another. For many cancer types including PC, it was also suggested that stromal cells may play a pivotal role in EMT initiation via a close crosstalk with tumour cells [11]. Last but not least, further level of complexity is reached with the description of partial EMT phenotypes, which may be an even more widespread and important phenomenon in cancer [9].
Due to the importance of metastasis in pathophysiology of cancer disease and mortality associated with it, there is a need to find novel factors that inhibit tumour cell invasion. Because of the regulation of cellular signalling, and on the basis of the results obtained in previous work [12], kinetin riboside seems to be a promising factor in this search.
N6-furfuryladenine, also known as kinetin (K), was described in 1955 as the first compound from the group of cytokinins—natural substances produced by plants known for their role in promoting proliferation and possibly differentiation of cells. Kinetin riboside (KR), in turn, is a ribosylated form of the former. Both compounds are thought to be generated in vivo in a reaction between hydroxyl radical and DNA following modified nucleotides excision. It was also suggested that this reaction could serve as a protective mechanism for cells to scavenge furfural generated upon DNA oxidation stress [13]. For years, a large number of research indicated biological activity of the aforementioned compounds in animals. Interestingly, while antiaging, proproliferative, antioxidative properties were attributed to cytokinins [14], respective ribosilated forms were shown to possess rather opposing effects leading to induction of DNA damage response genes, increased ROS generation and ATP depletion, growth arrest, and apoptosis [15,16,17,18]. Importantly, the latest was proved in some papers to be at least partially specific to cancer cells [19]. Although K and KR are mainly described as plant-derived substances, recent evidence raised a possibility that they could be generated in humans as K was shown to be present in lung cancer patients’ urine at levels 100-fold greater than in healthy subjects’ samples suggesting an increased generation of K in the first group [20]. Importantly, that supports the idea of close relations between cancer and K, as well as its derivatives.
In this study, we aim to elucidate the influence of KR treatment on PC cell lines in vitro, especially in the context of EMT traits and AI.
Methods
Cell culture
Human prostate cell lines (American Type Culture Collection—ATCC, Manassas, Virginia, USA)—PZHPV-7 and RWPE-1 (both derived from normal prostate peripheral zone epithelial cells), WPMY-1 (derived from stroma of normal prostate) as well as human prostate cancer cell lines (ATCC)—LNCaP (androgen-dependent cell line derived from lymph nodes metastasis), PC-3 (androgen-independent cell line derived from bone metastasis) and Du-145 (androgen-independent cell line derived from brain metastasis). Cells were cultured in prepared media as follows: for non-cancerous PZHPV-7 and RWPE-1 in Keratinocyte Serum Free Medium (K-SFM) enriched with BPE (0.05 mg/ml) and EGF (5 ng/ml), for WPMY-1 in Dulbecco's Modified Eagle's Medium with 5% FBS and for cancerous cell lines in RPMI-1640 with the addition of 10% FBS according to previously described procedure [12].
Primary cell line
The prostate tissue samples used in the study were surgically resected specimens (Department of Urology, Jagiellonian University Medical College, Krakow, Poland) from patients with diagnosed and histopathologically confirmed (Department of Pathology, Jagiellonian University, Medical College, Krakow, Poland [Consent of the Bioethics Committee])) as normal prostate tissue (n = 11; age 72 ± 10) or prostate cancer (CA, G1 n = 6, G2 n = 9, G3 n = 11, G4 n = 9; age 64 ± 10). The preparation of tissue slices was performed as previously described [21]. Twelve of the obtained tissue specimens showed satisfactory growth of the released cells and as a result five primary cell lines: normal (N), G1, G2, G3, and G4 (obtained from tissues with different grading of prostate cancer according to Gleason score) were derived. Kinetin riboside (KR) solutions in indicated concentrations were prepared as described previously [12], KR (Sigma) was dissolved in water to the concentration of 1 mM. Cells were treated with prepared dilutions of KR at final concentration of 2.5 or 5 μM in full growth media for given amounts of time for 48 or 72 h. Non-treated cells were used as a negative control. Cell culturing and stimulation were conducted according to previously described procedure [22].
Cytotoxixity of KR
Cells were seeded in triplicate into 96-well plate in the amount of 1.5 × 103 per well and incubated in the presence of KR at different evaluated concentrations and cultured for 48 and 72 h in standard conditions. Cytotoxicity was evaluated based on colorimetric measurement according to manufacturer’s instruction (Cytotoxicity Detection Kit, Roche Diagnostics, Poland) and previously described procedure [23]. Absorbance was measured at 492 nm by ELISA reader (Synergy HT, BIO-TEK, USA). No cytotoxic effect was noted for evaluated concentrations.
Cell proliferation
Cells were seeded in triplicate into 96-well plate in the amount of 1.5 × 103 per well and incubated with or without evaluated stimulants at different concentrations. ELISA BrdU (Roche) colorimetric immunoassay tests were performed according to manufacturer’s protocol as previously described [23]. Cell proliferation was evaluated based on absorbance measurement at λ = 450 nm performed with the use of ELISA plate reader (Synergy HT, BIO-TEK, USA).
Western blot
After incubation with indicated factors cells were harvested in lysis buffer (0.0625 M Tris/HCl pH: 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol) and total protein concentration was evaluated according to Pedersen protocol. Lysates containing equal amounts of protein were separated in 10% SDS-PAGE gel and transferred onto PVDF membrane. For protein detection specific primary antibodies against: AR, p-AR(Ser210/213), E-cad, p-Ecad(Ser838/Ser840), AKT, p-AKT(Ser 473), PTEN, p-PTEN(Ser380), RAS, RAF, ERK1/2, BAX, BAD, BCL-2, SNAIL, TWIST, ZEB1 and β-actin (Sigma-Aldrich, Poland) as for referral protein were used. Secondary rabbit or mouse antibodies conjugated with HRP (1:10,000, Cell Signaling Technology, Inc.) were utilised for detection. All steps were performed as previously described [22]. In order to obtain quantitative results, the bands (representing each data point) were densitometrically scanned using SynGene Gene Tools version 4.03.0 (Synoptics Ltd Beacon House, Nuffield Road Cambridge, CB4 1TF, UK). β-actin was used as a control. Protein level within the control group was arbitrarily set as 1. The procedure was performed as previously described [24].
RNA isolation, cDNA synthesis and detection
Total amount of RNA was isolated using RNeasy Plus Mini kit (Qiagen, Hilden, Germany) according to manufacturer’s instruction. The amount of total RNA was assessed in spectrophotometric measurement (NanoDrop Technologies, Wilmington, DE, USA). cDNA synthesis was conducted with the use of oligo(dT) starters and GoScript Transcriptase (Promega GmbH, Germany) according to provided protocol. Prepared cDNA was utilised for the assessment of gene expression at the mRNA level utilising Color OptiTaq PCR Master Mix (2×) (EURx, Poland) according to manufacturer’s instruction. Measurements were performed as previously described [22]. For primers used in the study PCR conditions were applied as follows: initial denaturation at 95 °C for 5 min; followed by 30 cycles: 30 s at 95 °C, 30 s at 58 °C (45 s at 55 °C for AR) and 30 s at 72 °C; and final extension at 72 °C for 10 min. Electrophoresis in the 1.5% agarose gel containing ethidium bromide was used for PCR product visualisation. Bands were normalised using GAPDH. Utilised primers’ sequences are presented in Supplementary Materials (Table S1). Primers were manufactured by Sigma-Aldrich (St. Louis, MO, USA). Fluorescence signal of ethidium bromide was detected by Bio-Rad ChemiDoc™ XRS + System (Bio-Rad, Hercules, CA, USA).
Real-time (RQ-PCR) analysis
RQ-PCR was performed using 7500 Real-Time PCR System (Applied Biosystems, Foster City, USA) and TaqMan probes. Specific primers and probes for AR, BAD, BAX, BCL-2, Snail, ZEB1 and ABL (control) genes were purchased from TIB MOLBIOL (Poznan, Poland) and Applied Biosystems (Cheshire, UK). RQ-PCR protocols were optimised for each studied gene according to the specific annealing temperatures (GAPDH—59 °C, 30 s; AR—55 °C, 45 s; Bax—49 °C, 30 s; Bcl-2—64 °C, 30 s; Snail 58 °C, 45 s; Zeb 58 °C, 40 s). RQ-PCR reactions and visualisation were carried out as previously described [22].
Cell migration and invasiveness
Cell migration potential and invasiveness were assessed utilising standard Boyden transwell method (BD BioCoat Tumor Invasion System No. 354166, Poland) according to manufacturer’s instruction. For quantification of migrating cells post-invasion cell labelling with calcein was performed and fluorescence of invading cells was evaluated.
Statistical analysis
All data concerning proliferation, cytotoxicity or cell invasion experiments represent the average of five wells in each experiment (n = 5). Data were expressed as mean ± standard deviation. In cases of RT-PCR and Western blot, each analysis was performed in triplicate. The significance of the differences between mean values were calculated using Student's t test or one-way analysis of variance and appropriate post hoc test (the ANOVA Kruskal–Wallis test). Differences are shown as significant at *p < 0.05, **p < 0.01 or ***p < 0.001, as indicated.
Results
KR decreases proliferation of prostate cells depending on malignancy potential
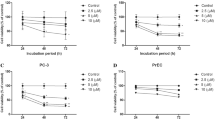
The influence of KR treatment on PC cell proliferation was assessed. Three different cell lines derived from normal prostate gland—RWPE-1, WPMY-1 and PZHPV, as well as three cell lines derived from PC with different characterisation—LNCaP (androgen dependent, low malignancy potential), Du145 (androgen independent, intermediate malignancy potential) and PC-3 (androgen independent, high malignancy potential) were used. Cells were treated with the indicated concentrations of KR for 48 or 72 h, and proliferation was assessed as described in “Methods” section. As shown in Fig. 1a, KR is capable of inhibiting prostate cell proliferation, which is dose- and time-dependent, at both concentrations. Also, a different response for KR treatment was observed for different cell types. The highest proliferation inhibition occurred in Du-145 and PC-3, whereas in non-carcinoid cell lines (RWPE-1, WPMY-1 and PZHPV) the effects were significantly weaker.

Proliferation of a malignant (LNCaP, PC-3, Du145) and non-malignant (PZ-HPV-7, RWPE-1, WPMY-1) human prostate cell lines and b primary cells—cancer G1, G2, G3, G4 and normal. Cells were treated with 2.5 μM and 5 μM Kinetin Riboside (KR). The cells were cultured as indicated: in typical medium (control) or treated with KR solution for 48 h and 72 h. The details are described in M&M. The results are given as the percentage of the control proliferation. Data represent mean values ± SD from three independent experiments. The significance was calculated using Student's t test or one-way analysis of variance and appropriate post hoc test (the ANOVA Kruskal–Wallis test). Differences are shown as significant at *p < 0.05, **p < 0.01 or ***p < 0.001
To further analyse the dependency of KR mediated proliferation inhibition on malignancy potential, the effect of KR treatment on primary cell lines prepared and classified as described in “Methods” section was assessed. Similarly to previous experiments, KR inhibited proliferation of prostate tissues in a time- and dose-dependent manner (Fig. 1b). KR diminished the proliferation to a greater extent in cells derived from tissues classified with higher Gleason score (G-score).
KR diminishes molecular traits of malignancy potential in prostate cancer cell lines
To elucidate molecular changes caused by KR treatment, PC cell lines (LNCaP, PC3, Du145, WPMY, RWPE and PZHPV) were treated with KR at concentration of 2,5 or 5 μM for 48 and 72 h. Protein expression at mRNA and protein levels was assessed as described in “Methods” section. Results from WB and RT-PCR are presented in Figs. 2, 3, 4, and 5.

Effect of KR on proliferation, migration and EMT markers in normal and cancer prostate cell lines (a) and primary cell lines (b): BAX, BCL-2, MMP-2, MMP-9, ZEB, SNAIL, TWIST and AR expression at mRNA level were determined by reverse transcription-PCR after incubation with typical medium (control) or 5 μM KR for 48 h. The upper lanes show the levels house-keeping gene GAPDH under the same culture conditions. Presented pictures are representatives of at least three independent experiments with comparable results

The effect of KR in human prostate cell lines on a the apoptotic pathway regulated by members of Bcl-2 family (Bax, Bad, Bcl-2); b the MAPK pathway (Ras, c-Raf, Erk1/2); c the EMT markers in transitional prostate cells (Snail, Twist, ZEB-1). Cells were treated with typical medium (control), 2.5 μM or 5 μM KR, as indicated and harvested after 72 h. β-actin was used as a protein loading control. Densitometry was used to normalise each proteins to β-actin protein level. Presented pictures are representative membranes of at least three independent experiments with comparable results

The effect of kinetin riboside (KR) on a malignant (LNCaP, PC-3, Du145) human prostate cell lines: Western blot analysis for Akt, p-Akt(Ser 473), PTEN, p-PTEN(Ser380). b Non-malignant (PZ-HPV-7, RWPE-1, WPMY-1) human prostate cell lines: western blot analysis for AR and p-AR(Ser210/213). Cells were treated as indicated in “Methods” section and harvested after 72 h. Densitometry was used to normalise Akt, PTEN or AR to β-actin protein level, and phosphorylated Akt, PTEN or AR to total Akt, PTEN or AR and to β-actin protein level. Presented pictures are representative membranes of at least three independent experiments with comparable results. The histograms represent densitometry data (mean ± SD). Values are denoted as **p < 0.01 or ***p < 0.001

a The effect of kinetin riboside (KR) on non-malignant (PZ-HPV-7, RWPE-1, WPMY-1) human prostate cell lines. Figure represents results of Western blot analysis for Akt, p-Akt(Ser 473), PTEN, p-PTEN(Ser380). Cells were treated as indicated in “Methods” section and harvested after 72 h. Densitometry was used to normalise Akt or PTEN to β-actin protein level, and phosphorylated Akt or PTEN to total Akt or PTEN and to β-actin protein level. Presented pictures are representative membranes of at least three independent experiments with comparable results. The histograms represent densitometry data (mean ± SD). Values are denoted as **p < 0.01 or ***p < 0.001. b Western blot analysis for E-cad and p-E-cad(Ser838/Ser840). Androgen-dependent LNCaP cell lines were treated as indicated in “Methods” section and harvested after 72 h. Densitometry was used to normalise E-cad to β-actin protein level, and phosphorylated E-cad to total E-cad and to β-actin protein level. Presented pictures are representative membranes of at least three independent experiments with comparable results. The histograms represent densitometry data (mean ± SD) of three independent experiments. Values are denoted as **p < 0.01 or ***p < 0.001
No full-length AR (fl-AR) expression in androgen insensitive cell lines (PC3 and Du145) was indicated (data not shown). In androgen-sensitive cell line LNCaP, the KR efficiently diminished the expression of AR at both mRNA and protein levels, which were, dose dependent. Moreover, decreased phosphorylation of AR at Ser 210/213 was observed after KR treatment.
A decrease of Akt protein level in each cell line was indicated, with the most prominent one in PC-3, and less visible in Du145 and WPMY-1. The protein level of Akt phosphorylated at Ser 473 was assessed and found to be significantly decreased after KR treatment in each cell line. The effect on phosphorylation was the most prominent in Du145. Furthermore, the protein level of potent Akt phosphatase PTEN was increased in Du145, WPMY and RWPE, but not in LNCaP and PC3, as those cell lines are PTEN negative [25]. Additionally, PTEN phosphorylation at Ser 380 was enhanced by KR.
Next, Ras/Raf/EKR pathway proteins expression in PC-3, Du145, LNCaP and PZHPV was examined; KR treatment was found to have caused a decrease in each protein level, which corresponded with the decrease in respective mRNA amount.
Real-time PCR experiment was performed; it confirmed KR capability of diminishing the expression of AR and BCL-2, as well as of increasing BAD and BAX expression in prostate cell lines, which was more evident in cancerous cells (Fig. 6a, c).

Activation of either the EMT or apoptotic pathways after treatment the cells with KR 5 µM for 72 h: a RQ-PCR analysis of AR, Snail, ZEB and b BAD, BAX, BCL-2 transcripts. To confirm the specificity of the PCR products, melting curve analysis was performed. The measurements were performed in triplicates according to the standard 2−ΔΔCT method with GAPDH used as a calibrator. All primer pairs were initially validated by a passing test for equal amplification efficiencies. The amplification efficiency was close to 2. Statistically significant difference from the corresponding control are marked as follows: *p < 0.05, **p < 0.005 or ***p < 0.001. c Global caspase activity—apoptosis pathway. The rate of apoptosis was estimated by caspase activity assay following 48-h incubation as indicated. The data represent the mean of three independent experiments, expressed as the percentage of control activity. Significantly different from the corresponding control are marked as follows: **p < 0.01, ***p < 0.001. d Migration through Matrigel-coated Boyden chamber. Representative results of at least three independent experiments are presented
Additionally, we evaluated the protein level of E-cadherin and E-cadherin phosphorylated at Ser838/840 in androgen-dependent LNCaP cell line, and showed that KR treatment increased both total protein amount and its phosphorylation.
KR affects apoptosis in PC via Bcl-2 family proteins
Our previous work showed that KR is a potent apoptosis activator [12]. The present results showing that KR was efficient in dose-dependent caspase activation in PC cell lines (Fig. 6a) are consistent with the previous reports. Caspases activation was predominantly present in cancerous cells. As shown in Figs. 3a and 4a, this effect is at least partly dependent on dysregulation of Bcl-2 family proteins’ level. KR was efficient in lowering protein and mRNA level of antiapoptotic Bcl-2, while subsequently increasing it in case of proapoptotic Bad and Bax. This influence on Bcl-2 and Bad was most prominent in tumorigenic Du145 and PC3 cell lines, and less evident in LNCaP and non-tumorigenic PZHPV.
KR reduces protein levels of EMT-associated transcription factors
A major hallmark and one of the substantial changes during EMT is the up-regulations of EMT-associated TFs—Snail, Twist1, and Zeb. To elucidate the influence of KR on molecular pathways governing EMT, its effect on the expression of those TFs was assessed. The results presented in Fig. 6b for protein amount and Fig. 2a for corresponding mRNA levels prove that KR is efficient in decreasing expression of Snail, Twist, and Zeb in each of the tumorigenic cell lines PC3, LNCaP, and Du145. Interestingly, no significant changes in normal PZHPV cell line were observed. These results were confirmed by real-time PCR experiment for Snail and ZEB (Fig. 6a).
KR inhibits migration potential of prostate cancer cell lines
As far as EMT is suggested to be implicated in increasing cellular motility and invasiveness, especially in case of cancer cells, we aimed to assess the influence of KR on migration utilising Boyden transwell method as described in “Methods” section. Results are presented in Fig. 6d. Among untreated cells, migration was detected in case of PC-3 and Du145 (high migration rate), LNCaP (moderate migration rate) and PZHPV (very low migration rate). KR treatment efficiently decreased the amount of migrated cells in each cell line and this effect was dose dependent.
KR influence on molecular pathways correlates with G-score
For a better insight into effects of KR on PC, mRNA level changes in primary cell lines triggered by KR were assessed. PCR analysis proved that KR efficiently diminished expression of mRNA for Bcl-2, MMP-2, MMP-9, Zeb, Snail, and Twist1, and increased BAX mRNA amount. Apart from MMP-9 mRNA, these changes were more prominent in cells derived from tissues characterised with higher G-score (Fig. 2b). Importantly, Zeb and Twist mRNA amount correlated positively with G-score in untreated cells.
Discussion
Our results indicate that KR is a compound capable of efficient inhibition of PC cell proliferation, which is dose- and time-dependent. Interestingly, proliferation inhibition among cell lines in response to KR treatment is positively correlated with their malignancy potential. Furthermore, the results obtained for primary cell lines presented in Fig. 1b support this notion—KR diminished proliferation to a greater extent in cells derived from tissues classified with higher Gleason score (G-score) which correlates with malignancy. That feature of KR is unusual for other carcinostatics.
To elucidate the mechanism of KR action, we screened changes in the expression of proteins known for their importance in PC biology upon treatment with this compound. First, we assessed androgen receptor (AR). As mentioned, AR is important predominantly in the early phases of prostate tumorigenesis, and with cancer progression it is often lost or changed, which may be advantageous for cancer cells. As expected, we found no full-length AR (fl-AR) expression in androgen insensitive cell lines characterised with high malignancy potential (PC3 and Du145; data not shown); however, the existence of AR variants (vAR) important in cancer progression in those cell lines [3] must be mentioned. In turn, the observed decrease of AR protein level in androgen-sensitive LNCaP cell line upon KR treatment may be partially responsible for proliferation inhibition in this cell line. Additionally, KR diminished phosphorylation of AR at Ser 210/213. This phosphorylation is thought to modulate AR mediated gene expression in a way beneficial for PC development, and its decrease may be attributed to inhibition of Akt known to target this site in AR to suppress AR-induced apoptosis [6].
Another protein thought to play an important role in PC progression, beside AR, is Akt. Akt signalling up-regulation is often a compensational event for androgen signalling loss at late stages of cancer development or during androgen deprivation therapy when it confers resistance towards this therapy. This pathway hyperactivity strongly correlates with malignancy potential [3, 5]. We proved KR to decrease total protein expression of Akt. Moreover, KR is highly efficient in inhibition of Akt activating phosphorylation at Ser 473, which is most evident in Du145. We indicated the increase in protein level of potent Akt phosphatase PTEN upon KR treatment. However, this may only partially explain Akt inhibition as PTEN is up-regulated only in Du145, WPMY and RWPE, but not in LNCaP and PC3, as those cell lines are PTEN negative [25]. Moreover, the inactivating phosphorylation of PTEN at Ser 380 is enhanced by KR treatment. Interestingly this modification of PTEN was reported to subsequently increase its stability and inactivate its phosphatase activity [26]. This is consistent with the observed increase in PTEN total protein level. On the other hand, the influence on enzymatic activity stays in opposition to the effects of KR on Akt. Moreover, as far as PC-3 is PTEN negative and yet most potent for KR mediated proliferation inhibition, it is unlikely that KR effects are mainly due to PTEN regulation. MAPK pathway with Ras, Raf, and Erk 1/2 being the most researched members, is the branch of cellular signalling that converges most proliferative signalling, thus its dysregulation plays an important role in PC [5]. It is activated by many growth factors and other proproliferative cues. KR efficiently decreased the amount of Ras, Raf, and Erk 1/2 protein level in PC cell lines thus diminishing proliferative signalling transmission. Again, this effect was slighter in PC-3, which is consistent with its highest malignancy potential, and suggests deeper dysregulation in molecular pathways. It would also be beneficial to assess the phosphorylation status of MAPK pathway in response to KR in the future.
Poor correlation between the influence on Akt or MAPK pathways and the excess of antiproliferative effect among cell lines suggest those may not be crucial molecular targets for KR carcinostatic activity. Alternatively, cell lines characterised with higher malignancy potential may be more dependent on those pathways, thus their inhibition may exert more prominent effects.
Importantly AR, MAPK, and Akt pathways are interconnected via a complex crosslinking. Crosstalk between them is multidirectional and context-dependent. For instance, Akt was shown to enhance or diminish transcriptional activity of AR, which was context-specific and dependent on the evaluated target gene [27].
As mentioned above, EMT has been recently extensively studied as a process involved in PC progression conferring metastatic potential, therapy resistance, and associated with poor prognosis. Although a precise primary mechanism underlying EMT induction is still elusive, many molecular pathways are known to enhance this process. Among them, Akt and MAPK signalling seem to play an important role in PC [8]. Although AR itself was proved to enhance EMT-associated TFs (Snail, Twist, Zeb) expression, androgen deprivation is also a known factor promoting EMT, possibly through accompanying compensatory up-regulation of Akt and MAPK signalling [8]. The obtained results clearly indicate the involvement of TFs such as Snail, Twiest and Zeb in the regulation of EMT. In fact, the TFs are crucial for EMT to occur. The best -recognised EMT molecular hallmark is E- to N-cadherin switch. Other changes involve enhanced matrix metalloproteinases (MMPs) expression, which enables cancer cells to invade surrounding tissues, thus conferring migratory and metastatic potential [9]. EMT-associated TFs, N-cadherin, and MMPs each may act in positive feedback loops activating other signalling pathways beneficial for cancer cells. It is important to note that EMT seems to be positively correlated with malignancy potential in case of PC [28]. Finally, it is noteworthy that EMT is not a binary process with many intermediate states between epithelial and mesenchymal differentiation that in fact may be most beneficial for cancer survival and development. Molecular phenomena leading to these transient states are similar to those leading directly to EMT [9].
In our work, we proved that KR efficiently inhibited EMT TFs expression in all researched PC cell lines, hence potentially inhibiting EMT.
Furthermore, KR inhibited migration of cells as assessed in Boyden chamber. This may be associated with the decreased motility resulting from EMT inhibition as well as the observed decreased MMPs expression after KR treatment. Interestingly, we did not observe any significant changes in EMT TFs and MMPs mRNA levels in normal PZHPV cell line, possibly because of its non-tumorigenic character resulting non-advanced EMT changes and lack of EMT activating signalling potent for inhibition.
Our results suggest that KR diminishes signalling through PI3K/Akt, MAPK, and AR in PC, although to a different extent in each cell line. Interestingly, in PC-3, which is characterised with the highest malignancy potential, proliferation inhibition by KR was the most marked, although decrease in PI3K/Akt, MAPK, and AR pathways were not most evident. This suggests the primary target of KR to be different from the aforementioned pathways at least in PC-3. On the other hand, Snail, Twist, and Zeb were down-regulated in each cell line in response to KR, and the magnitude of this effect was positively correlated with malignancy potential and G-score, thus potentially explaining why KR exerts stronger antiproliferative activity in those cells. It is possible that at least in PC-3 EMT inhibition by KRs occurs upstream of other researched molecular changes, which are secondary to disruption of positive feedback loops formed due to EMT programme. This may explain why the antiproliferative effect of KR is greatest in cells characterised by the highest malignancy potential and most advanced EMT-like molecular. It is possible that up-regulation of EMT TFs is pivotal for PC development towards more malignant states, and during this process cancer cells grow more and more dependent on their activity.
Additionally, we showed KR to increase E-cadherin and phosphorylated E-cadherin (Ser838/Ser840) protein levels in LNCaP cell line. Increased E-cadherin amount may be the hallmark of EMT reversion by KR and is itself linked to reduced invasiveness and migratory potential. Furthermore, enhanced phosphorylation at Ser838/Ser840 was shown to strengthen cell–cell adhesion and increase E-cadherin/β-Catenin interaction, thus decreasing WNT oncogenic signalling in tumour [29].
The KR influence on Bcl-2 family protein highlights the proapoptotic potential of this compound, which is consistent with our previous results. Interestingly, although KR universally causes an increase in proapoptotic Bax and Bak levels with subsequent decrease in antiapoptotic Bcl-2, the relative magnitude of these effects is different in each cell line. This is consistent with many reports that EMT TFs are able to modulate Bcl-2 family protein expression. Interestingly, it was also shown that Bcl-2 is not only up-regulated by EMT TFs, but its overexpression itself may induce EMT [30].
Our work highlights the efficacy of KR in EMT inhibition and, consequently, lowering metastatic potential. However, precise molecular mechanisms behind these phenomena are still elusive. Recently, KR was shown to act as neosubstrate for some kinases due to its similarity to ATP, which is possibly playing a physiological role in DNA damage response [31]. Off note, other compounds sharing structural similarity with KR are also known to act as distinct kinases inhibitors [32,33,34,35]. Consistently, KR itself is a potent inhibitor of DNPH1—an enzyme suggested to play a role in tumorigenesis [36]. It is possible that KR also directly affects kinase activity in case of EMT inhibition. Finally, we can say that KR as well as other cytokinins can work in a similar way as antiproliferative effect was previously attributed to ROS generation, ATP depletion, differentiation induction and Bcl-2/Bax balance modulation [19, 37]. On the other hand, kinetin itself is a potent antioxidant. Interestingly, kinetin was also reported to alter lipid bilayer properties [38] and to modulate mitochondrial respiration as observed by increase in transmembrane potential [39], which is in opposition to KR mediated effect [19]. Given the aforementioned importance of ROS balance in EMT development, it is possible that these phenomena also play a role in its inhibition.
Other possible mechanisms comprise cdk1 and 2 inhibition, as well as AMPK activation, TRAF6 activity modulation with following inhibition of Akt, TAK1 inhibition, and decreasing FPPS activity resulting in diminished Ras signalling [32,33,34,35]. Finally, although KR was proved not to be incorporated into DNA or RNA [14], it is possible that its biological activity may depend on direct interaction with transcriptional machinery due to its chemical similarity to other nucleosides.
References
Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov). Cancer Stat Facts: Prostate Cancer. 2019. https://seer.cancer.gov/statfacts/html/prost.html. Accessed 12 Jul 2019.
Parker C, Gillessen S, Heidenreich A, Horwich A. Cancer of the prostate: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015. https://doi.org/10.1093/annonc/mdv222.
Dai C, Heemers H, Sharifi N. Androgen signaling in prostate cancer. Cold Spring Harb Perspect Med. 2017. https://doi.org/10.1101/cshperspect.a030452.
Crawford E. Challenges in the management of prostate cancer. Br J Urol. 1992. https://doi.org/10.1111/j.1464-410x.1992.tb15865.x.
Saraon P, Jarvi K, Diamandis E. Molecular alterations during progression of prostate cancer to androgen independence. Clin Chem. 2011. https://doi.org/10.1373/clinchem.2011.165977.
Wen S, Niu Y, Lee S, Chang C. Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat Rev. 2014. https://doi.org/10.1016/j.ctrv.2013.07.008.
Yu P, Duan X, Cheng Y, Liu C, Chen Y, Liu W, Yin B, Wang X, Tao Z. Androgen-independent LNCaP cells are a subline of LNCaP cells with a more aggressive phenotype and androgen suppresses their growth by inducing cell cycle arrest at the G1 phase. Int J Mol Med. 2017. https://doi.org/10.3892/ijmm.2017.3125.
Montanari M, Rossetti S, Cavaliere C, Daniello C, Malzone MG, Vanacore D, Difranco R, Lamantia E, Iovane G, Piscitelli R, Muscariello R, Berretta M, Perdonà S, Muto P, Botti B, Bianchi AAM, Veneziani BM, Facchini G. Epithelial–mesenchymal transition in prostate cancer: an overview. Oncotarget. 2017; https://doi.org/10.18632/oncotarget.15686
Nieto M, Huang R, Jackson R, Thiery J. EMT: 2016. Cell. 2016. https://doi.org/10.1016/j.cell.2016.06.028.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014. https://doi.org/10.1038/nrm3758.
Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, Chiarugi P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial–mesenchymal transition and cancer stemness. Cancer Res. 2010. https://doi.org/10.1158/0008-5472.can-10-0785.
Dudzik P, Dulińska-Litewka J, Wyszko E, Jędrychowska P, Opałka M, Barciszewski J, Laidler P. Effects of kinetin riboside on proliferation and proapoptotic activities in human normal and cancer cell lines. J Cell Biochem. 2014. https://doi.org/10.1002/jcb.24991.
Barciszewski J, Rattan S, Siboska G, Clark B. Kinetin: 45 years on. Plant Sci. 1999. https://doi.org/10.1016/s0168-9452(99)00116-8.
Barciszewski J, Massino F, Clark B. Kinetin: a multiactive molecule. Int J Biol Macromol. 2007. https://doi.org/10.1016/j.ijbiomac.2006.06.024.
Ishii Y, Hori Y, Sakai S, Honma Y. Control of differentiation and apoptosis of human myeloid leukemia cells by cytokinins and cytokinin nucleosides, plant redifferentiation-inducing hormones. Cell Growth Differ. 2002;13(1):19–26.
Cabello C, Bair W, Ley S, Lamore S, Azimian S, Wondrak G. The experimental chemotherapeutic N6-furfuryladenosine (kinetin-riboside) induces rapid ATP depletion, genotoxic stress, and CDKN1A (p21) upregulation in human cancer cell lines. Biochem Pharmacol. 2009. https://doi.org/10.1016/j.bcp.2008.12.002.
Rajabi M, Gorincioi E, Santaniello E. Antiproliferative activity of kinetin riboside on HCT-15 colon cancer cell line. Nucleosides Nucleotides Nucleic Acids. 2012. https://doi.org/10.1080/15257770.2012.681825.
Cheong J, Goh D, Wan Hong Yong J, Ngin Tan S, Shi Ong E. Inhibitory effect of kinetin riboside in human heptamoa, HepG2. Mol BioSyst. 2009. https://doi.org/10.1080/15257770.2012.681825.
Choi B, Kim W, Wang Q, Kim D, Hong Yong JW, Kim K, Yoon HS. Kinetin riboside preferentially induces apoptosis by modulating Bcl-2 family proteins and caspase-3 in cancer cells. Cancer Lett. 2008. https://doi.org/10.1016/j.canlet.2007.11.014.
Barciszewski J, Mielcarek M, Stobiecki M, Siboska G, Clark B. Identification of 6-furfuryladenine (kinetin) in human urine. Biochem Biophys Res Commun. 2000. https://doi.org/10.1006/bbrc.2000.3928.
Laidler P, Dulińska J, Mrozicki S. Does the inhibition of c-myc expression mediate the anti-tumor activity of PPAR’s ligands in prostate cancer cell lines? Arch Biochem Biophys. 2007. https://doi.org/10.1016/j.abb.2007.03.013.
Dulinska-Litewka J, McCubrey JA, Laidler P. Increased Akt signaling resulting from the loss of androgen responsiveness in prostate cancer. Curr Med Chem. 2013;20(1):144–57.
Dulińska J, Gil D, Zagajewski J, Hartwich K, Bodzioch M, Dembińska-Kieć A, Langmann T, Schmitz G, Laidler P. Different effect of beta-carotene on proliferation of prostate cancer cells. Biochem Biophys Acta. 2005. https://doi.org/10.1016/j.bbadis.2004.12.004.
Górowska-Wójtowicz E, Hejmej A, Kamińska A, Pardyak L, Kotula-Balak M, Dulińska-Litewka J, Laidler P, Bilińska B. Anti-androgen 2-hydroxyflutamide modulates cadherin, catenin and androgen receptor phosphorylation in androgen-sensitive LNCaP and androgen-independent PC3 prostate cancer cell lines acting via PI3K/Akt and MAPK/ERK1/2 pathways. Toxicol In Vitro. 2017. https://doi.org/10.1016/j.tiv.2017.01.019.
Lotan T, Gurel B, Sutcliffe S, Esopi D, Liu W, Xu J, Hicks JL, Park BH, Humphreys E, Partin AW, Han M, Netto GJ, Isaacs WB, De Marzo AM. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clin Cancer Res. 2011. https://doi.org/10.1158/1078-0432.ccr-11-1244.
Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000. https://doi.org/10.1128/mcb.20.14.5010-5018.2000.
Koryakina Y, Ta H, Gioeli D. Androgen receptor phosphorylation: biological context and functional consequences. Endocr Relat Cancer. 2014. https://doi.org/10.1530/erc-13-0472.
Jędroszka D, Orzechowska M, Hamouz R, Górniak K, Bednarek A. Markers of epithelial-to-mesenchymal transition reflect tumor biology according to patient age and Gleason score in prostate cancer. PLoS ONE ONE. 2017. https://doi.org/10.1371/journal.pone.0188842.
Lickert H, Bauer A, Kemler R, Stappert J. Casein kinase II phosphorylation of E-cadherin Increases E-cadherin/β-catenin interaction and strengthens cell-cell adhesion. J Biol Chem. 2000. https://doi.org/10.1074/jbc.275.7.5090.
Kim E, Jung C, Song J, Park J, Um H. Pro-apoptotic Bax promotes mesenchymal-epithelial transition by binding to respiratory complex-I and antagonizing the malignant actions of pro-survival Bcl-2 proteins. Cancer Lett. 2018. https://doi.org/10.1016/j.canlet.2018.03.033.
Maiuri T, Bowie L, Truant R. DNA repair signaling of Huntingtin: the next link between late-onset neurodegenerative disease and oxidative DNA damage. DNA Cell Biol. 2019. https://doi.org/10.1089/dna.2018.4476.
Li M, Qi Y, Wei J, Lu L, Zhao X, Zhou L. N6-isopentenyladenosine promoted HeLa cell apoptosis through inhibitions of AKT and transforming growth factor β-activated kinase 1 activation. Tumor Biol. 2017. https://doi.org/10.1177/1010428317695966.
Scrima M, Lauro G, Grimaldi M, Di Marino S, Tosco A, Picardi P, Gazzerro P, Riccio R, Novellino E, Bifulco M, Bifulco G, D’Ursi AM. Structural evidence of N6-isopentenyladenosine as a new ligand of farnesyl pyrophosphate synthase. J Med Chem. 2014. https://doi.org/10.1021/jm500869x.
Pisanti S, Picardi P, Ciaglia E, Margarucci L, Ronca R, Giacomini A, Malfitano AM, Casapullo A, Laezza C, Gazzerro P, Bifulco M. Antiangiogenic effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, mediated by AMPK activation. FASEB J. 2014. https://doi.org/10.1096/fj.13-238238.
Vermeulen K, Strnad M, Kryštof V, Havlícěk L, Van der AA, Lenjou M, Nijs G, Rodrigus I, Stockman B, van Onckelen H, Van Bockstaele DR, Berneman ZN. Antiproliferative effect of plant cytokinin analogues with an inhibitory activity on cyclin-dependent kinases. Leukemia. 2002. https://doi.org/10.1038/sj.leu.2402378.
Amiable C, Pochet S, Padilla A, Labesse G, Kaminski P. N6-substituted AMPs inhibit mammalian deoxynucleotide N-hydrolase DNPH1. PLoS ONE ONE. 2013. https://doi.org/10.1371/journal.pone.0080755.
Zhang Z, Zou J, Huang Y, Wu L. Kinetin inhibits proliferation of hepatic stellate cells by interrupting cell cycle and induces apoptosis by down-regulating ratio of Bcl-2/Bax. J Huazhong Univ Sci Technol. 2015. https://doi.org/10.1007/s11596-015-1488-0.
Stillwell W, Hester P. Kinetin increases water permeability of phosphatidylcholine lipid bilayers. Plant Physiol. 1983. https://doi.org/10.1104/pp.71.3.524.
Liu Z, Bushnell W, Brambl R. Pontentiometric cyanine dyes are sensitive probes for mitochondria in intact plant cells. Plant Physiol. 1987. https://doi.org/10.1104/pp.84.4.1385.
Funding
This work was supported by MNISZW Grant: K/ZDS/005888; UJCM.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dulińska-Litewka, J., Gąsiorkiewicz, B., Litewka, A. et al. Could the kinetin riboside be used to inhibit human prostate cell epithelial–mesenchymal transition?. Med Oncol 37, 17 (2020). https://doi.org/10.1007/s12032-020-1338-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-020-1338-1