
Abstract
Bictegravir is a new integrase strand transfer inhibitor (INSTI) with a high genetic barrier to the development of HIV-1 resistance. The drug is co-formulated with the nucleos(t)ide reverse transcriptase inhibitors emtricitabine and tenofovir alafenamide (AF) in a single-tablet regimen (STR) for the once-daily treatment of HIV-1 infection in adults (bictegravir/emtricitabine/tenofovir AF; Biktarvy®). In phase 3 trials, bictegravir/emtricitabine/tenofovir AF was noninferior to dolutegravir-based therapy (dolutegravir/abacavir/lamivudine or dolutegravir plus emtricitabine/tenofovir AF) in establishing virological suppression in treatment-naïve adults through 96 weeks’ treatment and, similarly, was noninferior to ongoing dolutegravir/abacavir/lamivudine or boosted elvitegravir- or protease inhibitor (PI)-based therapy in preventing virological rebound over 48 weeks in treatment-experienced patients. No resistance emerged to any of the antiretrovirals in the STR. Bictegravir/emtricitabine/tenofovir AF is generally well tolerated, requires no prior HLA-B*5701 testing (making it more suitable for ‘rapid start’ treatment), fulfils the antiretroviral regimen requirement for patients with hepatitis B virus (HBV) co-infection (i.e. contains tenofovir AF and emtricitabine, both of which are active against HBV) and can be used in renally impaired patients with creatinine clearance (CRCL) ≥ 30 mL/min. Thus, although cost-effectiveness analyses would be beneficial, current data indicate that bictegravir/emtricitabine/tenofovir AF is a convenient initial and subsequent treatment option for adults with HIV-1 infection, including those co-infected with HBV, and provides the first non-pharmacologically boosted, INSTI-based, triple-combination STR suitable for patients with CRCL 30–50 mL/min.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
First unboosted, INSTI-based, triple-combination STR suitable for patients with CRCL 30–50 mL/min |
Noninferior to dolutegravir-based regimens in establishing virological suppression in treatment-naïve adults |
Maintains virological suppression in adults switched from a dolutegravir- or boosted elvitegravir- or PI-based regimen |
Generally well tolerated |
1 Introduction
Antiretroviral therapy (ART) has radically improved outcomes for patients with HIV-1 infection, with most patients now able to achieve virological suppression owing to the potency, tolerability and convenience of the available antiretroviral agents [1]. A combination of three active agents from at least two different classes is standard in realizing this goal, with regimens usually comprising two nucleos(t)ide reverse transcriptase inhibitors (NRTIs) plus either an integrase strand transfer inhibitor (INSTI), a non-nucleoside reverse transcriptase inhibitor (NNRTI) or a pharmacologically-boosted protease inhibitor (PI) [1,2,3,4], with INSTIs being the preferred first-line choice in the most recent treatment guidelines [1, 3, 4].
Until recently, the INSTI class included only raltegravir and elvitegravir (the limitations of which include a lower genetic barrier to resistance than some other agents and considerable cross resistance) and the second-generation INSTI dolutegravir (which has a more favourable resistance profile) [1, 5]. These INSTIs are available as single-agent formulations and/or single-tablet regimens (STRs) [6, 7], the latter of which are convenient, easy to take and have potential adherence advantages [1]. These STRs generally comprise an INSTI plus two NRTIs (e.g. dolutegravir/abacavir/lamivudine), with those containing elvitegravir also including the pharmacological boosting agent cobicistat [i.e. elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide (tenofovir AF) or tenofovir disoproxil fumarate (tenofovir DF)]. However, the drug interaction potential of cobicistat and/or the tolerability issues associated with some of the NRTI components (namely hypersensitivity and increased cardiovascular risk with abacavir, and renal and bone toxicity with tenofovir DF) [1], can limit the use of these STRs.
Recently, INSTI options have been expanded with the approval of a once-daily STR containing a new INSTI, bictegravir. This STR (Biktarvy®) combines bictegravir 50 mg with the NRTIs emtricitabine 200 mg and tenofovir AF 25 mg and is hereafter referred to as bictegravir/emtricitabine/tenofovir AF. This article reviews pharmacological, therapeutic and tolerability data relevant to the use of bictegravir/emtricitabine/tenofovir AF in adults infected with HIV-1.
2 Pharmacodynamic Properties
The pharmacodynamics of bictegravir are the main focus of this section, with the established pharmacodynamic profiles of emtricitabine and tenofovir AF discussed only briefly.
2.1 Antiviral Activity
Bictegravir was highly active against laboratory and clinical isolates of HIV-1 in vitro [50% effective concentration (EC50) of 0.02–6.6 nmol/L [8, 9]; protein-adjusted 95% effective concentration against laboratory strains of 361 nmol/L] [8, 10], displaying activity against HIV-1 isolates of all groups (M, N and O) and subtypes A–G (EC50 < 0.05–1.71 nmol/L) [8, 10, 11]. Notably, bictegravir, like the earlier second-generation INSTI dolutegravir, was significantly (p < 0.05) more active than the first-generation INSTIs elvitegravir and raltegravir against recombinant HIV-1 strains derived from INSTI-naïve patients [12].
Bictegravir also displayed in vitro activity against HIV-1 strains with mutations conferring resistance to elvitegravir and raltegravir, with EC50 values of < 5 nmol/L for all viruses with single INSTI resistance-associated mutations (RAMs) [e.g. Y143R, Q148R] and < 5 or < 10 nmol/L for the majority of those with double or triple INSTI RAMs (e.g. E92Q/N155H ± G163R) [8, 9, 13].
In this regard, bictegravir is similar to dolutegravir, although the overall activity profile of bictegravir against INSTI-resistant HIV-1 strains was slightly broader than that of dolutegravir in vitro [8, 9, 13]. In two studies, some mutant strains (14 of 57 [13] or 1 of 9 [9]) were significantly (p < 0.01) more susceptible to bictegravir than dolutegravir (e.g. G140S/Q148R [13] or G140S/Q148H [9], both of which reduce susceptibility to dolutegravir), whereas none [9] or numerically fewer (10 of 57) [13] strains were significantly more susceptible to dolutegravir. Moreover, across a panel of 47 patient-derived isolates with INSTI resistance, bictegravir was more potent than dolutegravir in terms of the overall mean fold-change in EC50 (FC; i.e. vs. wild-type virus) [2.8 vs. 5.8 nmol/L; p = 0.042] and the proportion of isolates that had an FC of ≥ 10 (2 vs. 17%) [8]. Bictegravir was at least twofold more potent than dolutegravir against thirteen of the isolates in this study (FCs for remaining isolates were similar for the two drugs) and significantly (p < 0.05) more potent against highly INSTI-resistant isolates carrying E92Q/N155H or G140C/S plus Q148R/H/K, with or without other INSTI RAMs [8]. Such differences in the activity profiles of bictegravir and dolutegravir may be due to greater flexibility in the bictegravir structure (allowing more conformational adaptation to overcome RAMs) [13] and slower dissociation of bictegravir from the integrase-DNA complex of G140S/Q148H variants [14].
Bictegravir was also fully active against HIV-1 variants resistant to other antiretrovirals, including NRTIs, NNRTIs and/or PIs, in vitro (FC 0.8–1.9) [8].
Highly synergistic activity against HIV-1 was seen with bictegravir when in combination with certain antiretrovirals, including emtricitabine or tenofovir AF [8]. Both emtricitabine and tenofovir AF are active against laboratory and clinical isolates of HIV-1; EC50 values range from 1 to 75 nmol/L for emtricitabine against subtypes A–G and from 0.1 to 12 nmol/L for tenofovir AF against all HIV-1 groups, including subtypes A–G [10, 11].
2.2 Resistance
Bictegravir, like dolutegravir, has a high barrier to resistance development in vitro [8, 15,16,17]. Cell culture resistance selection data suggest that at least two amino acid substitution pathways may be involved in conferring resistance to bictegravir (R263K/M50I and S153F plus transient T66I); however, the substitutions appear to be associated with only minimal reductions in susceptibility to the drug (≈ 3-fold for R263K/M50I, ≈ 2-fold for R263K or S153F, and 0.4- to 1.3-fold for M50I, T66I and T66I/S153F) [16]. Certain amino acid substitutions in the reverse transcriptase of HIV-1 can also reduce susceptibility to emtricitabine (M184V/I) and tenofovir AF (K65R, sometimes plus S68N or L429I; and transiently K70E) [10, 11], although selection of high-level resistance has not occurred with the latter drug over extended periods of culture in vitro [10].
Among treatment-naïve or -experienced adults with HIV-1 infection who received bictegravir/emtricitabine/tenofovir AF in five phase 3 clinical trials (Sect. 4) [18,19,20,21,22] and were eligible for resistance testing (n = 14), no treatment-emergent resistance (genotypic [10, 22] or phenotypic [10]) was evident to any of the STR antiretrovirals [10, 22], irrespective of HIV-1 subtype (B or non-B) or pre-existing resistance [23]. Findings were generally similar for corresponding recipients of the dolutegravir- or boosted PI- or INSTI-based comparator regimens in these trials [18,19,20,21,22], although a RAM developed in the reverse transcriptase in one recipient of a ritonavir-boosted PI plus dual NRTI regimen (L74V) [20] and one recipient of elvitegravir/cobicistat/emtricitabine/tenofovir AF (M184M/I/V) [22].
2.2.1 Cross Resistance
Certain combinations of RAMs that confer resistance to elvitegravir and raltegravir and/or reduce susceptibility to dolutegravir reduced the susceptibility of HIV-1 strains to bictegravir in vitro, including E138K/Q148K [8, 13], G140A/Q148K [13], L74M/G140A/Q148R [13], E138K/G140A/Q148K [13], T97A/G140S/Q148H [13, 24], L74M/T97A/G140S/Q148H [24] and G140S/Q148H/N155H [13] (EC50 values 12 to > 200 nmol/L where specified [8, 13]). Across 64 clinical isolates, susceptibility to bictegravir was reduced > 2.5-fold in 14 of the 24 isolates that carried G140A/C/S and Q148H/R/K, although most of the less susceptible isolates (9 of 14) also carried other RAMs (L74M, T97A or E138A/K) [10, 11]. Another in vitro study [24] supported these findings and demonstrated extensive cross-resistance between bictegravir and dolutegravir when G140S/Q148H was present in combination with other INSTI RAMs, including T97A and L74M. Reductions in bictegravir susceptibility have also been observed in site-directed HIV-1 strains with the dolutegravir and raltegravir RAM G118R, either alone (3.4-fold reduction) or in combination with T97A (2.8-fold reduction) [10, 11]. However, the clinical relevance of these in vitro data is not yet known.
Cross resistance amongst NRTIs is a well-established phenomenon. For instance, the emtricitabine RAM M184V/I confers resistance to lamivudine, and the tenofovir RAMs K65R and K70E reduce susceptibility to emtricitabine, as well as abacavir, didanosine and lamivudine [10]. Moreover, multi-NRTI-resistant strains of HIV-1 that carry a T69S double insertion mutation or a K65R-containing Q151M mutation complex are less susceptible to tenofovir AF [10, 11], as are strains with multiple thymidine analogue substitutions (M41L, D67N, K70R, L210W, T215F/Y, K219Q/E/N/R) [11].
2.3 Other Effects
Neither bictegravir (at up to six times the recommended dose) nor tenofovir AF (at up to five times the recommended dose) prolonged the PR or QT/corrected QT (QTc) interval in thorough QT/QTc studies in healthy volunteers; whether emtricitabine impacts the QT interval is unknown [11].
As with dolutegravir and some other antiretrovirals [4, 25], bictegravir can inhibit tubular secretion of creatinine, although any increases in serum creatinine levels that may occur with the drug are not indicative of actual glomerular filtration rate (GFR) changes and are thus not considered to be of clinical relevance [10].
3 Pharmacokinetic Properties
After oral administration of bictegravir/emtricitabine/tenofovir AF, with or without food, all components of the STR are readily absorbed, with the time to maximum plasma concentration being 2.0–4.0 h for bictegravir, 1.5–2.0 h for emtricitabine and 0.5–2.0 h for tenofovir AF [10, 11]. In intensive pharmacokinetic substudies (n = 15–28) of large phase 3 clinical trials (discussed in Sect. 4), patients receiving once-daily bictegravir/emtricitabine/tenofovir AF as recommended (Sect. 6) had mean bictegravir trough concentrations (2038–2576 ng/mL) that were 13- to 16-fold higher than the in vitro protein-adjusted effective concentration of the drug against wild-type HIV-1 [162 µg/mL (i.e. 361 nmol/L); Sect. 2]; moreover, the pharmacokinetic profiles of the other drugs in the STR were consistent with historical data in patients infected with HIV-1 [18,19,20,21].
The STR can be taken without regard to food [10, 11], as administering emtricitabine with food did not impact systemic exposure to the drug and taking bictegravir/emtricitabine/tenofovir AF or tenofovir AF with high/moderate fat meals caused modest, but clinically irrelevant, increases in bictegravir and tenofovir AF, respectively [10]. Plasma protein binding is high for bictegravir (> 99% in vitro) and tenofovir AF (≈ 80% ex vivo) and low for emtricitabine (< 4% in vitro) [10, 11] and the blood-to-plasma ratio of the respective drugs is 0.64, 1.0 and 0.6 [11].
Metabolism is the key route of elimination for bictegravir and tenofovir AF [10, 11]. Bictegravir is metabolized predominantly by CYP3A and UGT1A1, with each dose being eliminated primarily via the faeces (≈ 60%; as parent drug and oxidative metabolites) and to a lesser degree via the urine (35%; mainly as glucuronide and other oxidative metabolites/conjugates) [10]. Metabolism of tenofovir AF is via cathepsin A [in peripheral blood mononuclear cells (PBMCs)/macrophages] and carboxylesterase-1 (in hepatocytes), producing its major metabolite tenofovir, which undergoes phosphorylation to form the active moiety tenofovir diphosphate [10]. Elimination of tenofovir AF occurs after the drug is metabolized to tenofovir (which is excreted via glomerular filtration and active tubular secretion), with < 1% of a dose being eliminated as unchanged drug in the urine [10]. Emtricitabine undergoes limited metabolism [11] (via oxidation and glucuronidation) [10] and is eliminated primarily via glomerular filtration and active tubular secretion [11], with most of a dose being excreted in the urine (e.g. ≈ 86% [10]) and only ≈ 14% in the faeces [10, 11]. The median terminal plasma half-life is 17 h for bictegravir, 10 h for emtricitabine, 0.51 h for tenofovir AF [10, 11] and 32 h for tenofovir [10]; the half-life of tenofovir diphosphate within PBMCs is 150–180 h [11].
Bictegravir/emtricitabine/tenofovir AF can be used without dosage adjustment in patients with an estimated creatinine clearance (CRCL) of ≥ 30 L/min, although is not recommended for use in patients with CRCL < 30 mL/min [10, 11], as data are insufficient [10]. Similarly, mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment requires no adjustment of the bictegravir/emtricitabine/tenofovir AF dosage, although the STR is not recommended in patients with severe hepatic impairment (Child-Pugh class C) as it has not been assessed in this population [10, 11].
Race [10, 26], gender [10] and age [10] do not impact bictegravir, emtricitabine or tenofovir AF exposure to any clinically relevant extent according to population pharmacokinetic analyses [10] and a small pharmacokinetic study (n = 50) in Japanese and Caucasian subjects [26]; no dose adjustments are recommended [10, 11]. Bictegravir, emtricitabine and tenofovir AF pharmacokinetics have not been assessed in patients co-infected with hepatitis B virus (HBV) and/or hepatitis C virus (HCV) [11].
3.1 Drug Interactions
Given the role of CYP3A and UGT1A1 in bictegravir metabolism, plasma concentrations of the drug may decrease or increase if co-administered with potent inducers or inhibitors of these enzymes, respectively [10, 11]. In addition, both bictegravir and tenofovir AF are substrates of P-gp and BCRP; although the clinical relevance of this property is not yet known for bictegravir, co-administering bictegravir/emtricitabine/tenofovir AF with drugs that strongly impact these transporters may alter tenofovir AF absorption (e.g. inducers/inhibitors of P-gp may reduce/increase absorption and consequently plasma concentrations of tenofovir AF) [10]. Some drugs that induce CYP3A, UGT1A1 and/or P-gp are contraindicated for use in combination with bictegravir/emtricitabine/tenofovir AF, including rifampicin (EU [10]; USA [11]) and St John’s wort (EU [10]; not recommended in USA [11]). Some other inducers (e.g. rifabutin, rifapentine, carbamazepine, oxcarbazepine, phenobarbital, phenytoin) [10, 11] or inhibitors (atazanavir, cobicistat, azithromycin, clarithromycin, ciclosporin) [10] of these enzymes are also not recommended for use in combination with the STR or may require caution or the consideration of alternatives in the USA [11] and/or EU [10].
Bictegravir is an inhibitor of OCT2 and MATE1 [10, 11]; thus, plasma concentrations of drugs that are substrates of these transporters may increase upon co-administration with bictegravir/emtricitabine/tenofovir AF [11]. One such drug is dofetilide, which is contraindicated in combination with the STR in the USA due to the potential for serious/life-threatening events [11]. Another example is metformin, which requires risk/benefit consideration before being co-administered with bictegravir/emtricitabine/tenofovir AF in the USA [11] and monitoring/dosage adjustment if being co-administered with the STR in patients with renal impairment in the EU [10].
Bictegravir/emtricitabine/tenofovir AF should not be administered in conjunction with other antiretrovirals [10, 11] or adefovir dipivoxil (an HBV treatment) [10]. As with other INSTIs, in order to avoid bictegravir chelation, there are also administration timing adjustments and/or food requirements for the STR in patients taking medications/oral supplements containing polyvalent cations (such as magnesium, aluminium, iron [10, 11] or calcium [11]); however, sucralfate is not recommended for co-administration with the bictegravir STR in the EU [10].
Bictegravir and tenofovir AF do not inhibit/induce CYP or CYP3A enzymes, respectively, in vivo [10]. However, caution is advised in the EU if co-administering bictegravir/emtricitabine/tenofovir AF with methadone, as it is not yet possible to exclude the potential for bictegravir metabolites to inhibit CYP1A2, 2B6 and/or 2D6 [10]. Emtricitabine has a low potential for CYP-mediated drug interactions [10]. As emtricitabine and tenofovir AF elimination is largely renal, concentrations may be increased by drugs that reduce renal function or that also undergo active tubular excretion [10, 11].
4 Therapeutic Efficacy
This section reviews the clinical efficacy of bictegravir/emtricitabine/tenofovir AF in treatment-naïve (Sect. 4.1) or -experienced (Sect. 4.2) adults with HIV-1 infection, as evaluated in five randomized, active comparator-controlled, multicentre, phase 3, noninferiority studies of double-blind [18, 19, 21] or open-label [20, 22] design. The trials enrolled men and women (or just women [22]) with an estimated GFR (eGFR) of ≥ 30 [18] or ≥ 50 [19,20,21,22] mL/min; chronic HCV infection was permitted where specified [18, 20, 21]. Across trials, patients were predominantly men (83–90% in mixed-sex studies); some data are from abstracts/posters [22, 27, 28].
4.1 Treatment-Naïve Adults
The efficacy of bictegravir/emtricitabine/tenofovir AF in treatment-naïve adults has been compared with that of dolutegravir plus emtricitabine/tenofovir AF [18] and dolutegravir/abacavir/lamivudine [19] in two phase 3 trials. The studies enrolled patients with a viral load of ≥ 500 copies/mL and no resistance to any of the NRTI study drugs. One of the trials [19] also required patients to have a HLA-B*5701 and HBV negative status to avoid potential abacavir hypersensitivity or development of emtricitabine resistance (comparator regimen would not have provided adequate efficacy against HBV). Randomization of patients to study regimen was stratified by viral load, CD4+ cell count and geographical region. Across trials, patients had a median age of 31–34 years and most were asymptomatic (≈ 90%) [18, 19].
In terms of establishing virological suppression, the bictegravir/emtricitabine/tenofovir AF STR was noninferior to each of the dolutegravir-based regimens (dolutegravir/abacavir/lamivudine [19] and dolutegravir plus emtricitabine/tenofovir AF [18]), as evaluated by the proportion of full analysis set (FAS) patients who attained a viral load of < 50 copies/mL at 48 weeks (primary endpoint; Table 1). These findings were not influenced by baseline patient/disease characteristics (i.e. age, sex, race, viral load, CD4+ cell count, geographical region or study drug adherence) and were supported by those of per-protocol (PP) analyses, sensitivity analyses and other virological outcomes, including the proportion of patients who achieved a viral load of < 20 copies/mL and the change from baseline in viral load at 48 weeks (Table 1) [18, 19]. The greatest rate of HIV-1 RNA level decline was observed in the first 4 weeks of treatment, regardless of the study regimen [18, 19]. Consistent with these findings, improvements in CD4+ cell count at 48 weeks did not significantly differ between bictegravir/emtricitabine/tenofovir AF and dolutegravir-based therapy in either trial (Table 1) [18, 19].
Longer term, bictegravir/emtricitabine/tenofovir AF remained noninferior to both dolutegravir/abacavir/lamivudine [27] and dolutegravir plus emtricitabine/tenofovir AF [28] with regard to the proportion of FAS patients who had a viral load of < 50 copies/mL after 96 weeks of treatment (87.9 vs. 89.8%; 95% CI − 6.9, 3.1 [27]; 84.1 vs. 86.5%; 95% CI − 7.9, 3.2 [28]). PP analyses of this endpoint at 96 weeks were generally consistent with the FAS findings [27, 28]. The mean absolute CD4+ cell level at 96 weeks did not significantly differ between the treatments where specified [28].
When bothersome HIV-1 symptoms were assessed using the HIV-Symptom Index (HIV-SI) in one of these studies [19], some symptoms were significantly less frequent with bictegravir/emtricitabine/tenofovir AF than with dolutegravir/abacavir/lamivudine over 48 weeks’ treatment, with the most notable differences being in reports of fatigue/loss of energy, nausea/vomiting, dizziness/light-headedness and difficulty sleeping in adjusted logistic regression analyses; fatigue/loss of energy and nausea/vomiting were also among the symptoms that significantly favoured the bictegravir over the dolutegravir regimen in a longitudinal model [29].
4.2 Treatment-Experienced Adults
The efficacy of switching treatment-experienced adults (specifically women [22]) with virological suppression from their current ART regimen to bictegravir/emtricitabine/tenofovir AF has been evaluated in three phase 3 trials [20,21,22]. Patients must have maintained a viral load of < 50 copies/mL for ≥ 3 [21] or ≥ 6 [20, 22] months and have been on a stable ART regimen comprising a dual NRTI backbone plus either a PI (boosted atazanavir [20, 22] or darunavir [20]) or an INSTI (dolutegravir [21] or boosted elvitegravir [22]). Where specified, patients with resistance to the NRTI components [20, 21] or to dolutegravir [21] were excluded; one of the studies [21] also excluded patients with chronic HBV infection (as the comparator regimen would not have provided adequate efficacy against the virus). In the trial in which patients had baseline virological suppression with a dolutegravir-based regimen, the regimen consisted of dolutegravir, abacavir and lamivudine taken most commonly as an STR (Table 2) [21] and is thus hereafter referred to as dolutegravir/abacavir/lamivudine.
Switching to the bictegravir/emtricitabine/tenofovir AF STR was noninferior to remaining on a dolutegravir-based [21] or a boosted PI- [20, 22] or elvitegravir- [22] based triple ART regimen in terms of maintaining virological suppression, as measured by the proportion of FAS patients who experienced a viral load of ≥ 50 copies/mL (i.e. virological rebound) over 48 weeks of therapy (primary endpoint analysis; Table 2). Where reported [20, 21], evaluation of this endpoint in the PP population supported these findings. Moreover, the majority of patients in each treatment group had a viral load of < 50 or < 20 copies/mL at this timepoint [20,21,22], with no significant differences evident between the groups where specified [20, 21] (Table 2).
In subgroup analyses, the proportion of patients who had a viral load of < 50 copies/mL at 48 weeks did not significantly differ between the switched and unswitched groups, irrespective of patient/disease characteristics such as age [20, 21], race [20, 21], geographical region [20, 21], study drug adherence (< or ≥ 95%) [21] and sex [20, 21], with the latter finding supporting the trial conducted solely in women [22].
In terms of other endpoints, changes in CD4+ cell count at 48 weeks did not significantly differ between the switched and unswitched groups in one [20] of the two [20, 21] key studies that reported this outcome. The other trial reported a significant reduction in this parameter in patients who switched to bictegravir/emtricitabine/tenofovir AF versus those who remained on a dolutegravir-based regimen, although the difference did not remain significant after adjusting for baseline CD4+ cell counts (Table 2) [21].
In the study that compared bictegravir/emtricitabine/tenofovir AF with dolutegravir/abacavir/lamivudine [21], some HIV-SI-assessed bothersome HIV-1 symptoms were found to occur significantly less frequently with bictegravir/emtricitabine/tenofovir AF over 48 weeks’ treatment. These symptoms most notably included feeling nervous/anxious, nausea/vomiting, feeling sad/down/depressed and poor quality sleep in adjusted logistic regression analyses, with longitudinal analyses supporting these findings [29].
5 Tolerability
Treatment with bictegravir/emtricitabine/tenofovir AF for up to 96 weeks was generally well tolerated in treatment-naïve and—experienced adults with HIV-1 infection in the phase 3 trials discussed in Sect. 4 [18,19,20,21,22, 27, 28], with most adverse events (AEs) being mild or moderate in severity, where specified [18,19,20,21,22].
Over 48 weeks’ treatment, the tolerability profile of bictegravir/emtricitabine/tenofovir AF was more favourable than that of dolutegravir-based therapy (dolutegravir/abacavir/lamivudine [19, 21] or dolutegravir plus emtricitabine/tenofovir AF [18]) in terms of the incidence of treatment-related AEs (TRAEs), both in treatment-naive patients (18 vs. 26% [18]; 26 vs. 40% [19]) and in treatment-experienced patients (8 vs. 16% [21]), with the between-regimen difference being significant where specified (both p < 0.05) [18, 21]. The most common TRAEs with bictegravir/emtricitabine/tenofovir AF and dolutegravir-based therapy included headache (4 vs. 3%), diarrhoea (3 vs. 3%) and nausea (3 vs. 5%) in treatment-naïve patients [18] and headache (2 vs. 3%) in treatment-experienced patients [21]. Notably, the incidence of treatment-related nausea was significantly lower with bictegravir/emtricitabine/tenofovir AF than with dolutegravir/abacavir/lamivudine in each of these treatment settings (5 vs. 17% [19] and 0 vs. 2% [21]; each p ≤ 0.03), as was the incidence of flatulence in treatment-experienced patients (0 vs. 2%; p < 0.05) [21].
Over 48 weeks of treatment, few recipients of bictegravir/emtricitabine/tenofovir AF or dolutegravir-based therapy experienced serious TRAEs (0.3 vs. 0.3% [19]; 0.4 vs. 0% [21]) or discontinued therapy because of AEs (≤ 2.1 vs. ≤ 1.3%) [18, 19, 21], with no significant difference between the groups where specified [21]. Grade 3 or 4 treatment-emergent laboratory abnormalities did not markedly differ in incidence (15–17 vs. 11–15%) or nature between the bictegravir/emtricitabine/tenofovir AF and dolutegravir-based regimens [18, 19, 21], with increased creatine kinase (4 vs. 3% [19]; 3 vs. 2% [18]) and increased fasting low-density lipoprotein cholesterol (LDL-C) [2–5 vs. 3–5%] [18, 19, 21] being among the most common.
Longer-term data over 96 weeks of therapy were consistent with the 48-week findings, with bictegravir/emtricitabine/tenofovir AF being significantly (p < 0.05) more favourable than dolutegravir/abacavir/lamivudine [27] or dolutegravir plus emtricitabine/tenofovir AF [28] in the incidence of TRAEs (28 vs. 40% [27]; 20 vs. 28% [28]), including nausea (the most common TRAE; 6 vs. 17%) [27].
In contrast to these double-blind trials, all other comparisons [20, 22] were open label in nature. In one of these other comparisons, bictegravir/emtricitabine/tenofovir AF had a tolerability profile in treatment-experienced women generally similar to that of boosted INSTI- or PI-based regimens, of which elvitegravir/cobicistat/emtricitabine/tenofovir AF or DF was the most common (95% of patients) [22]. However, when compared solely with boosted PI-based therapy in the treatment-experienced setting, the incidence of TRAEs was 19% with bictegravir/emtricitabine/tenofovir AF and 2% with the comparator regimen, with this difference being driven predominantly by headache (5 vs. 0%), nausea, diarrhoea and flatulence (each 2 vs. 0%), although the open-label nature of the study may have impacted these findings (Sect. 7) [20]. Few patients in either group experienced serious TRAEs (0.3% of bictegravir/emtricitabine/tenofovir AF vs. 0% of boosted PI-based therapy recipients) or discontinued treatment because of AEs (0.7 vs. 0.3%). Grade 3 or 4 laboratory abnormalities occurred with a numerically lower incidence with bictegravir/emtricitabine/tenofovir AF than with boosted PI-based therapy (16 vs. 29%), due mainly to the between-group difference in adverse total bilirubin levels (1 vs. 15%) [20].
Patients co-infected with HIV-1 and HBV have experienced severe acute HBV exacerbations upon discontinuing emtricitabine- and/or tenofovir DF-containing products [11], and they may also experience such exacerbations after discontinuing bictegravir/emtricitabine/tenofovir AF [10, 11], because (like emtricitabine and tenofovir DF) tenofovir AF is active against HBV [1]. The EU [10] and US [11] prescribing information for bictegravir/emtricitabine/tenofovir AF include warnings/precautions to this effect (with the warning being boxed in the USA [11]) and recommend close clinical and laboratory monitoring for several months or more after discontinuing the STR in patients co-infected with HIV-1 and HBV. The prescribing information also carries various warnings and precautions relating to other AEs that have occurred/may occur with nucleos(t)ide analogues (e.g. lactic acidosis/severe hepatomegaly with steatosis [11] and mitochondrial dysfunction in exposed infants [10]) or ART in general (e.g. increased risk of severe/fatal hepatic adverse reactions in patients with chronic HBV or HCV infection [10]; immune reactivation/reconstitution syndrome [10, 11], opportunistic infections, osteonecrosis and increased bodyweight and levels of blood lipids and glucose [10]).
5.1 Renal Profile
The overall renal tolerability profile of bictegravir/emtricitabine/tenofovir AF was generally similar to that of dolutegravir-based regimens over 48 [18, 19, 21] or 96 [27, 28] weeks in treatment-naïve [18, 19, 27, 28] or –experienced [21] patients. There were no significant differences between bictegravir/emtricitabine/tenofovir AF and dolutegravir/abacavir/lamivudine with regard to median changes from baseline in proteinuria/tubular function measures [19, 21, 27] or serum creatinine [19] at these timepoints, where specified. Between-group differences in corresponding eGFR changes were either not significant (− 10.5 vs. − 10.8 mL/min [19]; − 6.9 vs. − 9.0 mL/min [30]) or significantly (p ≤ 0.01) favoured bictegravir/emtricitabine/tenofovir AF but were of no clinical relevance (− 7.8 vs. − 9.6 mL/min [27]; + 1.0 vs. − 1.8 mL/min [21]). Similarly, bictegravir/emtricitabine/tenofovir AF did not significantly differ from dolutegravir plus emtricitabine/tenofovir AF with regard to median changes from baseline in serum creatinine, although was associated with less extensive eGFR reductions (median change was − 7.3 vs. − 10.8 mL/min; p = 0.02) [18]. The observed regimen differences in eGFR may reflect tubular transporter affinity differences between bictegravir and dolutegravir [21] (e.g. bictegravir inhibits OCT2-mediated tubular secretion of creatinine to a lesser extent than dolutegravir [18]).
Bictegravir/emtricitabine/tenofovir AF did not significantly differ from a mixed treatment group of boosted elvitegravir- or PI-based regimens with regard to median changes in eGFR over 48 weeks in treatment-experienced women (− 1.8 vs. − 2.7 mL/min) [22]. However, when compared solely with boosted PI-based therapy in the treatment-experienced setting in another study [20], bictegravir/emtricitabine/tenofovir AF significantly reduced eGFR over 48 weeks (median changes from baseline were − 4.3 vs. + 0.2 mL/min; p = 0.0005), likely due to OCT2 or MATE1 transporter inhibition by bictegravir, although did not impact actual GFR (indicating the eGFR finding was of no clinical relevance). In each of these trials [20, 22], some measures of proteinuria/tubular function (urinary retinol-binding protein to creatinine ratio and urinary β2-microglobulin to creatinine ratio, but not urinary albumin to creatinine ratio) significantly (p < 0.001) improved after switching to the bictegravir STR versus remaining on comparator regimens that contained tenofovir DF. However, similar to the comparisons with dolutegravir/abacavir/lamivudine in treatment-naïve patients, there were no significant differences between bictegravir/emtricitabine/tenofovir AF and non-TDF-containing comparator regimens with regard to changes in proteinuria/tubular function measures [20, 22].
Across trials, there were no cases of proximal tubulopathy [19,20,21,22, 28], Fanconi syndrome [19,20,21, 28] or treatment being discontinued because of renal AEs [19, 21, 22, 28], and few (< 1%) bictegravir/emtricitabine/tenofovir AF recipients experienced serious renal AEs [11]. However, renal function should be monitored before and during treatment with bictegravir/emtricitabine/tenofovir AF in the USA, with discontinuation of the STR advised if Fanconi syndrome develops or renal function declines to a clinically relevant extent [11].
5.2 Lipid Profile
Over 48 weeks, lipid outcomes with bictegravir/emtricitabine/tenofovir AF were generally similar to those with dolutegravir-based therapy (dolutegravir/abacavir/lamivudine [19, 21] or dolutegravir plus emtricitabine/tenofovir AF [18]) in treatment-naïve [18, 19] and –experienced [21] patients. Among the fasting lipid parameters evaluated [total cholesterol (total-C), LDL-C, high-density lipoprotein cholesterol (HDL-C), triglycerides and total-C to HDL-C ratio], median changes in triglyceride levels (− 5 vs. + 3 mg/dL) [21] and total-C to HDL-C ratio (− 0.1 vs. − 0.2) [19] differed significantly (p < 0.05) between the bictegravir/emtricitabine/tenofovir AF and dolutegravir-based treatment groups in some trials, although the difference was not considered to be clinically relevant where specified [19]. Consistent with these findings, the proportion of patients who started lipid-lowering therapy generally did not significantly differ between the regimens, although was significantly lower with bictegravir/emtricitabine/tenofovir AF than with dolutegravir/abacavir/lamivudine in one study (1 vs. 4%; p = 0.033) [21]. Longer term, differences between bictegravir/emtricitabine/tenofovir AF and dolutegravir-based therapy in lipid changes after 96 weeks of therapy were either not significant [28] or were statistically significant but not considered to be clinically relevant (median changes in respective groups were + 15 vs. + 8 mg/dL for total-C, + 17 vs + 7 mg/dL for LDL-C and − 0.1 vs. − 0.2 for total-C to HDL-C ratio) [27].
Bictegravir/emtricitabine/tenofovir AF for 48 weeks in treatment-experienced patients also did not significantly differ from boosted PI-based therapy in terms of median changes from baseline in total-C, LDL-C or HDL-C levels or the proportion of patients who initiated lipid-lowering therapy, although significantly (p ≤ 0.033) improved triglyceride levels (median change − 6 vs. + 4 mg/dL) and the total-C to HDL-C ratio (median change − 0.2 vs. 0) [20]. These findings are supported by those of the women-only trial (that compared bictegravir/emtricitabine/tenofovir AF with boosted elvitegravir- or PI-based regimens) [22]. However, notably, the findings were dependent on the NRTIs within the PI-based regimen, as switching to bictegravir/emtricitabine/tenofovir AF from an emtricitabine/tenofovir DF-containing PI regimen had no impact on lipid profile whereas switching from an abacavir/lamivudine-containing PI regimen significantly (p < 0.05) improved levels of total-C, LDL-C and triglycerides and the total-C to HDL-C ratio [20].
5.3 Bone Profile
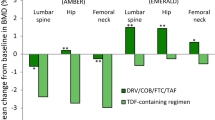
Over 48 weeks of treatment, mean changes from baseline in bone mineral density (BMD) with bictegravir/emtricitabine/tenofovir AF were small and did not significantly differ from those seen with dolutegravir/abacavir/lamivudine in treatment-naïve patients (− 0.8 vs. − 1.0% at hip; − 0.8 vs. − 0.6% at lumbar spine) [19] or treatment-experienced patients (+ 0.2 vs. + 0.3% at hip; + 0.7 vs. + 0.4% at lumbar spine) [21]. Where reported, fractures were uncommon in each of the respective treatment groups (2 vs. 3% of patients), with none considered to be treatment related or resulting in study drug discontinuation [21]. Longer term, differences between bictegravir/emtricitabine/tenofovir AF and dolutegravir/abacavir/lamivudine in terms of mean changes from baseline in BMD at the hip (− 1.1 vs. − 1.3%) or spine (− 0.7 vs. − 0.22%) remained non-significant after 96 weeks’ therapy in treatment-naïve patients [27].
6 Dosage and Administration
The bictegravir/emtricitabine/tenofovir AF STR is indicated in the EU for the treatment of HIV-1 infection in adults without evidence of viral resistance to the INSTI class, emtricitabine or tenofovir [10]. It is also indicated in the USA for the treatment of HIV-1 infection in adults with no ART history or to replace the current ART regimen in those who have had virological suppression (i.e. viral load < 50 copies/mL) for ≥ 3 months on stable ART, provided they have no history of treatment failure and no known RAMs to any of the STR components [11]. The recommended dosage of bictegravir/emtricitabine/tenofovir AF is one tablet (50/200/25 mg) administered orally once daily, with or without food [10, 11]. Being a complete ART regimen, bictegravir/emtricitabine/tenofovir AF should not be co-administered with any other antiretroviral agents [10, 11]. Local prescribing information should be consulted for detailed information regarding use in special patient populations, drug interactions, contraindications and other warnings and precautions.
7 Place in the Management of HIV-1 Infection
To support long-term successful management of HIV-1 infection, ART regimens should be selected/switched on the basis of factors such as strain resistance, patient comorbidities and regimen characteristics, including efficacy, tolerability, drug interaction potential and convenience [1, 2]. The latter aspect has improved considerably over the years with the growing availability of STRs (Table 3) and other fixed-dose antiretroviral combinations [7]. STRs require administration of only one tablet each day and thus make once-complex ART regimens simpler and easier for patients to take, which may improve adherence and consequently therapeutic outcomes [7]. Most STRs available to date provide standard triple-combination ART, i.e. two NRTIs plus either an NNRTI, a boosted PI or an INSTI (Table 3), with the most recent guidelines considering INSTIs to be the preferred third component for most treatment-naïve patients [1, 3, 4].
INSTIs are the most recent class of antiretrovirals for treating HIV-1 infection [6] and include the first-generation agents raltegravir and elvitegravir and the second-generation agents dolutegravir and bictegravir. Like dolutegravir [31], the latest INSTI bictegravir has a high genetic barrier to the development of resistance (Sect. 2.2), largely overcoming the relative resistance limitations of raltegravir and elvitegravir [5, 31]. In vitro, bictegravir demonstrates slightly broader activity against INSTI-resistant HIV-1 strains than dolutegravir and is active against some strains with reduced dolutegravir susceptibility (Sect. 2.1), although cross-resistance between the two INSTIs is evident (Sect. 2.2.1), the clinical relevance/implication of which remains to be determined.
Bictegravir is available as part of an STR in combination with emtricitabine plus tenofovir AF [10, 11], a recent tenofovir prodrug that has replaced tenofovir DF in the most recent STRs because of its improved bone and renal profile [7]. Approval of bictegravir/emtricitabine/tenofovir AF for the treatment for HIV-1 infection (Sect. 6) is supported by several pivotal phase 3 trials in which it was noninferior to dolutegravir-based regimens in establishing virological suppression over up to 96 weeks’ treatment in treatment-naïve adults (Sect. 4.1) and, similarly, switching to bictegravir/emtricitabine/tenofovir AF was noninferior to remaining on a dolutegravir-based or boosted elvitegravir- or PI-based regimen in preventing virological rebound over 48 weeks in treatment-experienced patients (Sect. 4.2). Importantly, no resistance emerged to any of the antiretrovirals in the STR in these studies, providing clinical support for the high resistance barrier observed with bictegravir in vitro (Sect. 2.2).
Bictegravir/emtricitabine/tenofovir AF was generally well tolerated in clinical trials (Sect. 5) and patients felt that they experienced certain bothersome symptoms of HIV-1 (including certain gastrointestinal and CNS symptoms) less frequently with bictegravir/emtricitabine/tenofovir AF than with dolutegravir/abacavir/lamivudine (Sects. 4.1 and 4.2). Bictegravir/emtricitabine/tenofovir AF was associated with fewer TRAEs than dolutegravir-based regimens (mainly due to better gastrointestinal tolerability) and more TRAEs than PI-based therapy (Sect. 5). However, interpretation of the latter finding requires consideration of the open-label design of the trial and the fact it compared switching to bictegravir/emtricitabine/tenofovir AF with remaining on a well-tolerated regimen, a phenomenon that has skewed the tolerability findings of other similarly-designed studies [32]. Although INSTIs are well-tolerated agents [1], their potential association with weight gain and obesity [33, 34] requires further investigation, as does the recent finding that dolutegravir may increase the risk of neural tube birth defects if taken during pregnancy [35].
Besides bictegravir/emtricitabine/tenofovir AF, other INSTIs are available in the form of single agents (dolutegravir, raltegravir) and/or STRs (Table 3) [1]. Most of these STRs comprise an INSTI plus two NRTIs, which negates the need for an NRTI backbone to be administered separately and thus makes them more convenient than single-agent INSTI formulations. However, cost and an inability to adjust drug dosages can limit STR use [2]. In addition to these constraints, dolutegravir/abacavir/lamivudine requires HLA-B*5701 status testing/confirmation (as patients with this allele can experience life-threatening hypersensitivity with abacavir [4]), making it unsuitable for ‘rapid start’ treatment, and there are also cardiovascular concerns with abacavir that need consideration [3]. By contrast, the elvitegravir STRs contain cobicistat (to pharmacologically boost elvitegravir) which has the drawback of increasing the likelihood of drug interactions [4]. Drug interaction potential is important to consider when selecting an ART regimen, given the need to manage comorbidities, such as metabolic disorders and co-infections (like tuberculosis) [2, 3].
For patients with HBV co-infection, ART regimens should include tenofovir AF or DF [1, 3], as these agents are active against HBV as well as HIV-1 [1]; emtricitabine and lamivudine also have activity against HBV, but do not provide adequate protection in the absence of other anti-HBV drugs [1]. The bictegravir- and elvitegravir-based STRs each fulfil the ART requirement for patients co-infected with HBV (i.e. contain tenofovir AF or DF), whereas lamivudine is the only active component against HBV within the dolutegravir-based triple-combination STR (Table 3). Patient HBV status must therefore be determined before initiating this dolutegravir STR [2], and an additional antiviral may be required if it is used in patients with HBV co-infection [36], which would increase pill burden and potentially also costs.
Bictegravir/emtricitabine/tenofovir AF is recommended in the most recent guidelines of the DHHS [1], EACS [3] and IAS-USA Panel [4] as one of the initial treatment options suitable for most individuals infected with HIV-1 (and as a ‘rapid start’ option by the IAS-USA [4]). The other ART options recommended by these bodies for use in this setting include the dolutegravir triple-combination STR (Table 3) [1, 3, 4] and dolutegravir in combination with emtricitabine plus tenofovir AF [1, 3, 4], with the DHSS [1] and EACS [3] also recommending dolutegravir in combination with emtricitabine plus tenofovir DF, and raltegravir in combination with emtricitabine plus tenofovir AF or DF. BHIVA guidelines generally provide similar INSTI recommendations [2]. However, given the more favourable bone and renal profile of tenofovir AF, the drug is preferred for certain patient groups, such as those with/at risk of renal disease [1,2,3,4], osteopenia [3] or osteoporosis [1,2,3], those who have had fragility fractures or tenofovir DF toxicity [3] and those taking nephrotoxic agents [3]. Notably, bictegravir/emtricitabine/tenofovir AF, like elvitegravir/cobicistat/emtricitabine/tenofovir AF, can be used in patients with CRCL > 30 mL/min, whereas the renal limitations for other INSTI-based triple-combination STRs are more extensive, with elvitegravir/cobicistat/emtricitabine/tenofovir DF limited to patients with CRCL ≥ 70 mL/min (because of potential renal toxicity with tenofovir DF) and dolutegravir/abacavir/lamivudine limited to patients with CRCL ≥ 50 mL/min (because renal impairment can increase lamivudine exposure).
In addition, bictegravir/emtricitabine/tenofovir AF has the smallest tablet size among the INSTI-based triple-combination STRs (Table 3), which some patients may prefer and/or find more acceptable. Another recently introduced INSTI-based STR, dolutegravir/rilpivirine, likewise has the benefit of a small tablet size (Table 3) and contains two, rather than three, antiretrovirals, which may help minimize antiretroviral exposure and thus long-term toxicity. However, this NRTI-sparing STR is limited to treatment-experienced patients with virological suppression for ≥ 6 months, must be taken with a meal, is contraindicated for use in combination with proton pump inhibitors [37, 38] and the long-term risk/benefit of dual (rather than triple) agent ART regimens remains to be determined.
Although cost-effectiveness analyses would be beneficial, current data indicate that bictegravir/emtricitabine/tenofovir AF is an effective, generally well-tolerated and convenient initial and subsequent treatment option for adults with HIV-1 infection, including those co-infected with HBV or requiring rapid ART initiation. It also provides the first non-pharmacologically boosted, INSTI-based, triple-combination STR suitable for use in patients with CRCL 30–50 mL/min.
Data Selection Bictegravir/Emtricitabine/Tenofovir Alafenamide: 124 records identified
Duplicates removed | 27 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 12 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 47 |
Cited efficacy/tolerability articles | 12 |
Cited articles not efficacy/tolerability | 26 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were Biktarvy, bictegravir/emtricitabine/tenofovir alafenamide. Records were limited to those in English language. Searches last updated 25 October 2018 | |
Change history
22 March 2019
The article Bictegravir/Emtricitabine/Tenofovir Alafenamide: A Review in HIV-1 Infection, written by Emma D. Deeks, was originally published Online First without open access. After publication in volume 78, issue 17, pages 1817���1828 Gilead Sciences requested that the article be Open Choice to make the article an open access publication. Post-publication open access was funded by Gilead Sciences. The article is forthwith distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made
22 March 2019
The article Bictegravir/Emtricitabine/Tenofovir Alafenamide: A Review in HIV-1 Infection, written by Emma D. Deeks, was originally published Online First without open access. After publication in volume 78, issue 17, pages 1817���1828 Gilead Sciences requested that the article be Open Choice to make the article an open access publication. Post-publication open access was funded by Gilead Sciences. The article is forthwith distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made
22 March 2019
The article Bictegravir/Emtricitabine/Tenofovir Alafenamide: A Review in HIV-1 Infection, written by Emma D. Deeks, was originally published Online First without open access. After publication in volume 78, issue 17, pages 1817���1828 Gilead Sciences requested that the article be Open Choice to make the article an open access publication. Post-publication open access was funded by Gilead Sciences. The article is forthwith distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made
References
Department of Health and Human Services. Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV. 2018. https://aidsinfo.nih.gov/guidelines. Accessed 30 Oct 2018.
British HIV Association. BHIVA guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy. 2015 (2016 interim update). http://www.bhiva.org. Accessed 30 Oct 2018.
European AIDS Clinical Society. Guidelines Version 9.1. 2018. http://eacsociety.org. Accessed 30 Oct 2018.
Saag MS, Benson CA, Gandhi RT, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2018 recommendations of the International Antiviral Society-USA Panel. JAMA. 2018;320(4):379–96.
Anstett K, Brenner B, Mesplede T, et al. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology. 2017;14(36):1–16.
Tang Q, Lu H. Latest advances in the efficacy, tolerability, and monotherapy of integrase inhibitors. Biosci Trends. 2017;11(4):490–5.
Sebaaly JC, Kelley D. Single-tablet regimens for the treatment of HIV-1 infection. Ann Pharmacother. 2017;51(4):332–44.
Tsiang M, Jones GS, Goldsmith J, et al. Antiviral activity of bictegravir (GS-9883), a novel potent HIV-1 integrase strand transfer inhibitor with an improved resistance profile. Antimicrob Agents Chemother. 2016;60(12):7086–97.
Hassounah SA, Alikhani A, Oliveira M, et al. Antiviral activity of bictegravir and cabotegravir against integrase inhibitor-resistant SIVmac239 and HIV-1. Antimicrob Agents Chemother. 2017;61(12):1–9.
Gilead Sciences. Biktarvy 50 mg/200 mg/25 mg film-coated tablets: EU summary of product characteristics. 2018. http://www.ema.europa.eu/. Accessed 30 Oct 2018.
Gilead Sciences. Biktarvy® (bictegravir, emtricitabine, and tenofovir alafenamide): US prescribing information. 2018. https://www.accessdata.fda.gov/. Accessed 30 Oct 2018.
Neogi U, Singh K, Aralaguppe SG, et al. Ex-vivo antiretroviral potency of newer integrase strand transfer inhibitors cabotegravir and bictegravir in HIV type 1 non-B subtypes. AIDS. 2018;32(4):469–76.
Smith SJ, Zhao XZ, Burke TR Jr, et al. Efficacies of cabotegravir and bictegravir against drug-resistant HIV-1 integrase mutants. Retrovirology. 2018;15(37):1–18.
White KL, Niedziela-Majka A, Novikov N, et al. Bictegravir dissociation half-life from HIV-1 G140s + Q148H integrase/DNA complexes [abstract no. 497]. Top Antivir Med. 2017;25(Suppl. 1):204s–5s.
Brenner BG, Oliveira M, Ibanescu RI, et al. In vitro selected resistance to new integrase inhibitors by B & non-B subtype viruses [abstract no. 549]. Top Antivir Med. 2018;26(Suppl. 1):233s.
US FDA. Application number: 210251Orig1s000 multi-discipline review. 2017. https://www.accessdata.fda.gov/. Accessed 30 Oct 2018.
Oliveira M, Ibanescu RI, Anstett K, et al. Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology. 2018;15(1):56.
Sax PE, Pozniak A, Montes ML, et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380-1490): a randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet. 2017;390(10107):2073–82.
Gallant J, Lazzarin A, Mills A, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV-1 infection (GS-US-380-1489): a double-blind, multicentre, phase 3, randomised controlled non-inferiority trial. Lancet. 2017;390(10107):2063–72.
Daar ES, De Jesus E, Ruane P, et al. Efficacy and safety of switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from boosted protease inhibitor-based regimens in virologically suppressed adults with HIV-1: 48 week results of a randomised, open-label, multicentre, phase 3, non-inferiority trial. Lancet HIV. 2018;5(7):e347–56.
Molina J-M, Ward D, Brar I, et al. Switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from dolutegravir plus abacavir and lamivudine in virologically suppressed adults with HIV-1: 48 week results of a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet HIV. 2018;5(7):e357–65.
Kityo C, Hagins D, Koenig E, et al. Switching to bictegravir/emtricitabine/tenofovir alafenamide in women [poster no. 500]. In: Conference on Retroviruses and Opportunistic Infections. 2018.
Acosta R, White K, Garner W, et al. HIV-1 subtype (B or non-B) had no impact on the efficacy of B/F/TAF or resistance development in five phase 3 treatment-naive or switch studies [abstract no. THPEB077]. In: 22nd International AIDS Conference. 2018.
Zhang WW, Cheung PK, Oliviera N, et al. Accumulation of multiple mutations in vivo confers cross-resistance to new and existing integrase inhibitors. J Infect Dis. 2018;218(11):1773–6.
Milburn J, Jones R, Levy JB. Renal effects of novel antiretroviral drugs. Nephrol Dial Transplant. 2017;32(3):434–9.
Zhang H, West S, Vu A, et al. A study evaluating the pharmacokinetics, safety, and tolerability of the bictegravir/emtricitabine/tenofovir alafenamide (B/F/TAF) single tablet regimen (STR) in Japanese subjects [abstract no. MOPEB0332]. In: 9th International AIDS Society Conference on HIV Science. 2017.
Wohl DA, Yazdanpanah Y, Baumgarten A, et al. A phase 3, randomized, controlled clinical trial of bictegravir in a fixed-dose combination, B/F/TAF, vs ABC/DTG/3TC in treatment-naïve adults at week 96 [abstract no. LB4]. In: IDWeek. 2018.
Stellbrink HJ, Arribas J, Stephens JL, et al. Phase III randomized, controlled clinical trial of bictegravir coformulated with FTC/TAF in a fixed-dose combination (B/F/TAF) versus dolutegravir (DTG) + F/TAF in treatment-naïve HIV-1 positive adults: week 96 [abstract plus presentation]. In: HIV Drug Therapy Conference. 2018.
Wohl D, Clarke A, Maggiolo F, et al. Patient-reported symptoms over 48 weeks among participants in randomized, double-blind, phase III non-inferiority trials of adults with HIV on co-formulated bictegravir, emtricitabine, and tenofovir alafenamide versus co-formulated abacavir, dolutegravir, and lamivudine. Patient. 2018;11(5):561–73.
Chuck S. Renal data at 96 weeks for study 1490. California: Gilead Sciences. 2018. (Data on file).
Yombi JC, Pozniak AL. Is it time for integrase inhibitors to be the preferred regimen for the first-line treatment of HIV-1-infected naive patients? AIDS Rev. 2016;18(2):89–100.
Llibre JM, Hung CC, Brinson C, et al. Efficacy, safety, and tolerability of dolutegravir-rilpivirine for the maintenance of virological suppression in adults with HIV-1: phase 3, randomised, non-inferiority SWORD-1 and SWORD-2 studies. Lancet. 2018;391(10123):839–49.
Norwood J, Turner M, Bofill C, et al. Weight gain in persons with HIV switched from efavirenz-based to integrase strand transfer inhibitor-based regimens. J Acquir Immune Defic Syndr. 2017;76(5):527–31.
Bakal DR, Coelho LE, Luz PM, et al. Obesity following ART initiation is common and influenced by both traditional and HIV-/ART-specific risk factors. J Antimicrob Chemother. 2018;73(8):2177–85.
European Medicines Agency. New study suggests risk of birth defects in babies born to women on HIV medicine dolutegravir [media release]. http://www.ema.europa.eu. Accessed 18 May 2018.
ViiV Healthcare UK Ltd. Triumeq 50 mg/600 mg/300 mg film-coated tablets: EU summary of product characteristics. 2018. http://www.ema.europa.eu/. Accessed 30 Oct 2018.
ViiV Healthcare UK Ltd. Juluca 50 mg/25 mg film-coated tablets: EU summary of product characteristics. 2018. http://www.ema.europa.eu/ema/. Accessed 30 Oct 2018.
ViiV Healthcare. Juluca (dolutegravir and rilpivirine) tablets, for oral use: US prescribing information. 2018. https://www.accessdata.fda.gov/. Accessed 30 Oct 2018.
Acknowledgments
During the peer review process, the manufacturer of bictegravir/emtricitabine/tenofovir AF was also offered the opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflicts of interest
Emma Deeks is a salaried employee of Adis/Springer, is responsible for the article content and declares no relevant conflicts of interest.
Additional information
The manuscript was reviewed by: D. M. Burger, Radboud University Medical Centre Nijmegen, Nijmegen, The Netherlands; C. Godfrey, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA; F. Maggiolo, Division of Infectious Diseases, ASST Papa Giovanni XXIII, Bergamo, Italy
The original version of this article was revised due to a retrospective Open Access request.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Deeks, E.D. Bictegravir/Emtricitabine/Tenofovir Alafenamide: A Review in HIV-1 Infection. Drugs 78, 1817–1828 (2018). https://doi.org/10.1007/s40265-018-1010-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-1010-7