
Abstract
The introduction in clinical practice of pharmaceutical products known as biosimilars, as part of a more complex series of progress in the field of biological drugs, represents an excellent therapeutic resource. A biosimilar drug is a biological/biotechnological drug that is highly similar to an approved reference biologic product. Given their complexity, biosimilars require attention and a continued vigilance to ensure appropriate use, especially in cancer therapy. There is the urgent need, both at Italian and European levels, of clear and more comprehensive guidelines to elucidate the open questions. Probably, the acquisition of new data, obtained from larger samples of patients than those used in the pre-approval studies and with extremely variable clinical conditions, will allow clarifying the extent to which biosimilar drugs are similar in safety and efficacy to their biologic reference drug. The aims of this article are to provide health professionals with basic, but essential information about biosimilars, and to identify current critical points and future perspectives for clinical practice, cancer care, regulatory aspects, and pharmacovigilance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A biosimilar is a biological/biotechnological drug that contains a version of the active substance of an already authorized original biological product (reference biologic product) in the European Economic Area (EEA). The biosimilar drug is similar to the reference biologic product in terms of quality characteristics, biological activity, safety, and efficacy [1].
Currently, in Europe a pharmaceutical patent lasts 20 years from the date the patent protection is issued and may be extended up to 5 additional years [2, 3]. The patents of some essential biological drugs in oncology, monoclonal antibodies (mAbs), have expired between the years 2013–2014 [4]. This aspect has opened the door to a competitive market, which should lead to lower prices of these drugs by approximately 20–30 % [5]. Indeed, the complexities in development and production of biological drugs lead to very high costs.
The potential of biosimilars are twofold: on the one hand, they represent a new option for the prevention and treatment of severe and debilitating diseases and, on the other, thanks to their improved economic sustainability on the European health systems, they ease patient access to innovative medicines.
The aims of this article is to provide health professionals with basic, but essential information about biosimilars, and to identify current critical points and future perspectives for clinical practice, cancer care, regulatory aspects, and pharmacovigilance. This article does not contain any new studies with human or animal subjects performed by any of the authors.
Similar but Not Identical
Biosimilar drugs, put on the market at reduced costs compared to the originator, are often considered as generic drugs of lower quality on the belief that a lower price indicates lower quality. That view is totally wrong. The difference between a generic and biosimilar is clear-cut, and development and regulatory pathways for marketing approval as biosimilar drug are even more rigorous than those of a generic drug (Table 1).
The concept of generic can be associated with the term bioequivalence. A drug is defined as generic when it is bioequivalent to the medicinal product from which it is derived, or rather:
-
Contains the same quantity and quality of active ingredients (however, the excipients may vary);
-
Presents the same dosage and method of administration;
-
Has equivalent bioavailability;
-
Produces the same clinical effects and, therefore, has the same therapeutic indications.
The generic drug is a copy of the ‘brand name’ product from which it is derived and whose patent is expired. The manufacturing of a generic drug is made by chemical synthesis through standardized and reproducible procedures, and it is the same as the originator drug. Consequently, the process of marketing authorization is simplified; the producer presents a dossier on a generic drug to the regulatory authority in charge of approving it (in Italy, Agenzia Italiana del Farmaco, AIFA); the dossier must contain the data proving the quality and bioequivalence of the generic drug.
The quantitative evaluation of bioequivalence is based on the pharmacokinetic parameters assessing drug absorption and spread throughout the body through the bloodstream. According to the guidelines issued by the European Medicines Agency (EMA) [6], 90 % of the confidence interval [derived from the ratio of the average values of the parameters C max (maximum serum concentration) and AUC (area under the curve)] must fall within the range of acceptability (80–125 %). However, the manufacturer of the drug is not required to repeat the studies on safety and effectiveness because these have already been proven by the reference medicine.
Conversely, for the biosimilar drug everything is based on biosimilarity. The biosimilar and its reference product must be comparable regarding quality, safety, and efficacy. The biosimilar, being similar but not identical to its originator, may differ both in form and in structure: It is obtained from irreproducible production processes subject to variability (the drug is obtained from living organisms, which are more complex than a simple chemical molecule) [7]. For biosimilars, the plasma production process markedly affects the safety profile and efficacy of the drug [8]. Moreover, the current legislation states that the manufacturer of a particular originator drug holds the patent on the techniques of production, and; therefore, there may be substantial differences between the biosimilar and its reference product.
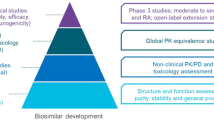
It is clear that the biosimilar is not a generic drug and, as such, cannot be approved by a simplified dossier [1]. In 2005, the EMA proposed guidelines for biosimilars [8]; the biosimilar must undergo a series of studies to compare it to its originator drug and to obtain marketing authorization. Together, these studies are defined as Comparability Exercise [1], which initially consists of studies on quality (biological and physic–chemical comparisons); then continues with comparative non-clinical studies (toxicity, pharmacokinetics, and pharmacodynamics), to end with clinical trials (efficacy and clinical safety, including immunogenicity). The aim is to demonstrate that, despite the intrinsic variability, the biosimilar has no significant clinical difference (of efficacy) compared to its reference product. The objective of developing a biosimilar drug is the similarity with the originator regarding efficacy, safety, and quality.
It should be noted that given the complex molecular structure of biological drugs (including biosimilars), every little change during its production, transport, and storage can result in significant differences in efficacy and clinical safety. Therefore, the regulatory authorities (AIFA and EMA) conduct rigorous inspections to ensure strict compliance with good manufacturing practice (GMP) and good distribution practice (GDP) [8].
Biosimilars in Oncology
In Europe, the first biosimilars to enter into clinical practice were erythropoiesis stimulating agents (ESAs) (e.g., epoetin alfa) and colony stimulating factors (CSFs) (e.g., filgrastim) [9]. These biosimilars are considered “first generation” biopharmaceuticals, and they are used in supportive care, for example, to reduce the occurrence of neutropenia associated with cancer treatment.
The mAbs drugs, an even more innovative category of biopharmaceuticals, has successfully entered oncologic clinical practice several years ago, proving useful in treatment of several cancers. These mAbs ‘second-generation’ biopharmaceuticals are large proteins, much more complex than the simple proteins present in first generation biopharmaceuticals, which are produced through genetic engineering. Consequently, the mAbs are to be considered in all aspects biotechnological medicinal products. Their anti-tumor activity is carried out on molecular targets, which are molecules or receptors located within the cell and are involved in the growth, angiogenesis, and cell proliferation. There are many advantages to using mAbs in oncology. First and foremost, these drugs can cause a considerable enhancement for chemotherapy and conventional therapies. Secondly, because of their improved selectivity against cancer cells, mAbs cause less toxicity to healthy cells, although side effects directly associated with their use are also present [10].
In 2013, the EMA approved Remsima® (Celltrion Healthcare Hungary Kft) and Inflectra® (Hospira UK Ltd.), which are biosimilars of infliximab, a mAb used for the treatment of inflammatory diseases (e.g., rheumatoid arthritis, Crohn’s disease, ulcerative colitis) [11].
Currently, in Europe there are no biosimilar mAbs available to treat neoplastic diseases; however, it is only a matter of time. Some of these drugs are already in the final stages of clinical trials. The following or already expired patent of many mAbs commonly used in the treatment of cancer (e.g., rituximab and trastuzumab), will make short-term biosimilar-based therapies possible [4].
In economically emerging countries such as India, China, and in South America, the production of biological drugs is on the rise. In fact, the need for these accessible and affordable lifesaving drugs has led these countries to add biosimilar monoclonal drugs to their trading market [9].
The extensive introduction of biosimilar drugs in economically emerging countries into clinical practices was favored by less stringent guidelines than European or American ones [12]. In India, for instance, Reditux® (Dr. Reddy’s Laboratories) is commonly used. Reditux is registered as a copy of the patented rituximab, a mAb for the treatment of non-Hodgkin lymphomas [13]. The approval pathway for Reditux in India followed a single-arm trial and no clinical head-to-head trials were made. This aspect has been widely discussed by Qureshi et al. [14]: the authors emphasize the fact that for biosimilars, similarity in efficacy and safety with the reference product must be demonstrated in adequately large head-to-head clinical trials. Consequently, not being able to apply the definition of ‘biosimilar drug’ by the EMA or by the US Food and Drug Administration (FDA) to the pharmaceuticals, Reditux cannot be considered a true biosimilar drug but rather a me-too drug. A me-too drug is manufactured following research pathways previously explored. It is not intended to be operated on comparability with the reference biological drug and, consequently, the registration process is conventional.
Biosimilars: The Need
The primary reason for the actual push for biosimilar drug use is the reduced cost compared to the originator. The cost reduction is of paramount importance, especially in oncology, where the expenditures associated with antineoplastic agents and immunomodulators are very high [15], affecting the sustainability of National Health Systems: for example, in Italy the costs of biosimilars are entirely borne by the Sistema Sanitario Nazionale (SSN) [16]. The introduction of biosimilar oncology drugs should have a future impact on the use of resources; it is believed that the biosimilars may be priced 20–30 % lower than their originators [5]. However, one must be aware that the price will vary and depend on the market, the competition between manufacturers and agreements of reimbursement between regulatory authorities and producers. It is unlikely that the cost reductions obtainable by biosimilars may be comparable to those seen in generic drugs. The development and the registration procedures of a biosimilar drug are just as long and complicated as those of a biological drug, and, therefore, expensive. In any case, the cost of a biosimilar is still less than the reference biological product [17], and this aspect fully justifies the introduction of this new class of drugs.
Critical Aspects of Biosimilar Oncology Drugs
Study Design
In oncology, the effects of therapy are defined as endpoints. Some endpoints relate to the disease and some relate to the patient. These are fundamental criteria in establishing the efficacy of a cancer treatment. The EMA, in the guidelines set for biosimilar mAbs, sets as primary endpoints: absence of disease; absence of disease progression; and overall survival. The goal of clinical trials that leads to the approval of a biosimilar is to demonstrate that there are no significant clinical differences with the reference product [18]; therefore, primary endpoints must be alike between the two drugs. In other words, therapeutic equivalence must be demonstrated, but it is not simple. In the absence of data, there is a lack of certainty concerning the equivalence between biosimilar and its reference; often skepticism or doubt arise in both patient and physician. To overcome this issue, a more accurate assessment of the endpoints would be required, which should be as much as possible calibrated on a specific biosimilar versus its originator in addition to more extensive clinical trials testing therapeutic equivalence or non-inferiority of the biosimilar.
Immunogenicity
One of the most important issues regarding biological medicine, and, thus, also biosimilars, is its immunogenic potential. Since most biological drugs are of a protein nature, the patient to whom the drug is given may develop antibodies because that protein is recognized as ‘non-self’ [19]. This aspect can affect the concentration of the drug in the blood and, therefore, its ability to produce a therapeutic effect. The immunogenicity can be a clinically insignificant event, but, even rarely, may cause serious adverse reactions: immunogenicity may cause hypersensitivity, anaphylaxis, infusion reactions, and loss of efficacy [8, 20]. Consequently, this must be evaluated case by case. The ability of biological drugs to induce immune responses may depend on several factors: the particular properties of the biological molecule, the patient characteristics, concomitant treatments, the administration mode or, finally, on changes introduced in production processes.
In this regard, one of the most sensational events occurred in the 1990s. The change made to the formulation of epoetin alfa for subcutaneous use caused pure red cell aplasia in different patients. This serious side effect was produced by replacing serum albumin with stabilizing agents (polysorbate 80 and glycine) [21].
The example demonstrates that even a small change can greatly affect the safety profile of a biological drug. Therefore, the regulatory authorities require a drug’s manufacturers to conduct studies under continuous monitoring of product safety, both before and after placing it on the market (so-called post-marketing studies) [22].
Interchangeability
In certain clinical situations, an oncologist may decide to replace one drug with another. This practice is defined as interchangeability. The interchangeability takes place when the two drugs are expected to be capable of producing the same clinical effect. The originator biological drugs and their biosimilars are similar, but not necessarily identical concerning efficacy and toxicity. Therefore, it cannot be assumed that they are automatically interchangeable [1].
Interchangeability is a process where the National Authority decides that drugs of the same class are interchangeable. The EMA do not address the issue of interchangeability for biosimilars, leaving to the medical provider the decision (and legal responsibility) to replace one drug with another. Both the EMA and the AIFA recommend therapeutic continuity for each patient already being treated; however, there is no reason not to prescribe directly biosimilar drugs to naïve patients, i.e., patients not previously treated.
The EMA approves biosimilar drugs based on all the same therapeutic indications of the originators, which, thus, allows extrapolating data from the studies conducted on the originator. Extrapolation in cancer therapy, however, requires extreme caution, as there is no data on long-term effects. Especially in the case of biosimilar mAbs used in oncology, a case-by-case assessment is required as well as a cautious attitude, in the spirit of “knowledge and belief.”
Substitutability
Substitutability is conceptually different from interchangeability. In Italy, the AIFA defines substitutability [1] as the replacement of a prescribed drug with a drug that has the following properties: usually more economical; bioequivalent, and with the same active ingredients, form, and dosage.
Substitutability is an active and/or passive process at the pharmacy level. In Italy, the replacement can be accomplished by the pharmacist and is allowed only between drugs included in the so-called “price transparency list” issued by the AIFA. Currently, only generic drugs and their originators appear in these lists. Therefore, the AIFA excludes the possibility of automatically replacing a biological medicine with the biosimilar [1]. Consequently, the choice to opt for therapy with a biosimilar drug or with its originator falls again on the medical provider. Since there are no standardized rules at the Italian national level, each Region has implemented individual measures, creating further confusion and inequalities in access to care [17].
Another aspect to consider is that the automatic replacement could generate misleading correlations between a drug and an adverse drug reaction (ADR), affecting the accuracy of data obtained from pharmacovigilance.
Nomenclature
Usually, the nomenclature of generic drugs refers to the international nonproprietary names (INN): the name of the generic drug is made by the active ingredient followed by the name of the manufacturer (e.g., olanzapine, Teva). For biosimilars assignments by the INN would not be entirely correct: the drug is not structurally identical to its originator, and the same INN name might associate products with different safety profiles [23].
The unique identification of a biosimilar is necessary for several reasons: first, to ensure the traceability of the medication; then, to avoid compromising pharmacovigilance and, above all, to ensure safety. In fact, the most frequent errors in therapy by the patient or healthcare provider are caused by the use of drugs that have similar names, graphics, or phonetics (Look Alike-Sound Alike, LASA).
Since 2006 the World Health Organization (WHO) has been looking for a name for biosimilars that is universal and more suitable than the INN names. In Europe, the choice of resorting to INN to name a biosimilar lies with the manufacturer, and there is not specific legislation outlining how the trade name should be assigned to a biological/biosimilar drug.
Pharmacovigilance
Post-marketing surveillance (an essential part of pharmacovigilance) is the set of interdisciplinary activities that aims to make the drug use as safe as possible, i.e., ensuring that the benefits of the drugs continue to outweigh the risks even after their marketing. The task of pharmacovigilance is to detect potential ADRs or changes in the frequency of adverse events, which are predictable and already known, and to implement, if necessary, specific preventive measures. Pharmacovigilance monitors, reports and evaluates all medications, even those that have been used for a long time or usually used as self-medication.
The biological and biosimilar drugs are considered a priority for the activities of the pharmacovigilance and, for this reason, the European Union has included them in the ‘List of Drugs Subject to Additional Monitoring.’ The identification of these drugs is by a black, inverted, equilateral triangle in the Illustrated leaflet and the summary of product characteristics (SPC) [24].
If there is a suspected ADR, the health professional must notify the National Pharmacovigilance Network through the completion of a report form. Since there may be differences between biological and biosimilar drugs, even between different batches of the same product, the report form must indicate the trade name of the drug and the lot number. This arrangement allows the product alleged to have caused an ADR to be easily traced [25].
Conclusions
Given their extreme complexity, biosimilar drugs must be used wisely in the treatment of cancer and with proper attention and awareness. Without a doubt, biosimilars represent an excellent therapeutic resource at an affordable cost that should not be underestimated.
At the European level, there is an urgent need for more clear and consistent guidelines to clarify open issues. Unfortunately, knowledge of these innovative drugs is still imprecise. The correct and comprehensive information on the use of biosimilars, especially in oncology, should be a priority on the part of the regulators, the media, and health professionals.
The acquisition of new data, obtained from larger samples of patients than those used in the pre-approval studies and with extremely variable clinical conditions, will allow clarifying the extent to which these drugs are similar in terms of safety and efficacy to their biologic reference drug. It is for this reason that all pharmacovigilance activities will provide a significant contribution to a deeper understanding of all aspects characterizing a biosimilar drug.
References
European Medicines Agency. Guideline on similar biological medicinal products. London. 2005. http://emea.europa.eu/pdfs/human/biosimilar/043704en.pdf. Accessed 13 July 2016.
Decreto Legislativo 10 febbraio 2005, n. 30. Codice della proprietà industriale, a norma dell’articolo 15 della legge 12 dicembre 2002, n. 273.Camera dei Deputati. 2005. http://www.camera.it/parlam/leggi/deleghe/05030dl.htm. Accessed 16 Apr 2016.
European Commission. Regolamento (CEE) n. 1768/92 del Consiglio, del 18 giugno 1992, sull’istituzione di un certificato protettivo complementare per i medicinali. 1992. http://eur-lex.europa.eu/eli/reg/1992/1768/oj. Accessed 14 Apr 2016.
Baldo P, De Paoli P. Pharmacovigilance in oncology: evaluation of current practice and future perspectives: pharmacovigilance in oncology and current practice. J Eval Clin Pract. 2014;20(5):559–69.
European Generic Medicines Association. Factors supporting a sustainable european biosimilar medicines market. Medicines for Europe. 2014. http://www.medicinesforeurope.com/?s=Factors+supporting+a+sustainable+european+biosimilar+medicines+market. Accessed 26 Apr 2016.
Committee for Medicinal Products for Human Use. Guideline on the investigation of bioequivalence. European Medicines Agency. 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed 26 Apr 2016.
Kathmann W. Biosimilars—Potenziale, Risiken und offene Fragen. Aktuelle Rheumatologie. 2014;39(02):80–81. https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-0034-1376214. Accessed 26 Apr 2016.
Associazione Italiana di Oncologia Medica. Farmaci biotecnologici e biosimilari. La rivoluzione dei biotech: dalle eritropoietine agli anticorpi monoclonali in oncologia. Associazione Italiana di Oncologia Medica. 2013. http://bio-similari.it/wp-content/uploads/2014/04/2013_AIOM-Farmaci_biotecnologici_e_biosimilari11.pdf. Accessed 26 Apr 2016.
Mellstedt H. Anti-neoplastic biosimilars—the same rules as for cytotoxic generics cannot be applied. Ann Oncol. 2013;24(Suppl 5):v23–8.
National Cancer Institute (NCI). Targeted cancer therapies. National Cancer Institute. 2014. http://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet. Accessed 24 Apr 2016.
European Medicines Agency. Press release: European Medicines Agency recommends approval of first two monoclonal-antibody biosimilar. 2013. http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2013/06/news_detail_001837.jsp&mid=WC0b01ac058004d5c1. Accessed 04 May 2016.
Kumar R, Singh J. Biosimilar drugs: current status. Int J Appl Basic Med Res. 2014; 4(2): 63–66. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4137643/.
Roy PS, John S, Karankal S, Kannan S, Pawaskar P, Gawande, J et al. Comparison of the efficacy and safety of Rituximab (Mabthera™) and its biosimilar (Reditux™) in diffuse large B-cell lymphoma patients treated with chemo-immunotherapy: a retrospective analysis. Indian J Med Paediatr Oncol. 2013;34(4):292–298. http://www.ncbi.nlm.nih.gov/pubmed/24604960.
Qureshi ZP, Magwood JS, Singh S, Bennet CL. Rituximab and biosimilars-equivalence and reciprocity. Biosimilars. 2013;(3):19–25. http://www.ncbi.nlm.nih.gov/pubmed/24829884.
Agenzia Italiana del Farmaco. L’uso dei Farmaci in Italia. Rapporto nazionale Osmed 2013. 2015. http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.655.1281&rep=rep1&type=pdf. Accessed 24 Apr 2016.
Ministero Della Salute. Decreto 4 aprile 2013. Criteri di individuazione degli scaglioni per la negoziazione automatica dei generici e dei biosimilari. Ministero della Salute. 2013. http://www.gazzettaufficiale.it/eli/id/2013/06/06/13A04795/sg. Accessed 24 Apr 2016.
Pani L. Farmaci Biotecnologici e biosimilari: Innovazione e Sostenibilità del sistema pubblico. Agenzia Italiana del Farmaco. 2013. www.senato.it/documenti/repository/…/Prof.%20PANI%20aifa.pdf.
Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products. European Medicines Agency. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf.
Bennet CL, Chen B, Hermanson T, Wyatt MD, Schulz RM, Georgantopoulos P, et al. Regulatory and clinical considerations for biosimilar oncology drugs. Lancet Oncol. 2014;15(13):e594–e605. http://www.ncbi.nlm.nih.gov/pubmed/25456378.
European Commission. What you need to know about biosimilar medicinal products. A consensus information document. European Commission. 2013. http://ec.europa.eu/DocsRoom/documents/8242/attachments/1/translations/en/renditions/native. Accessed 26 Apr 2016.
Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian J-J, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–475. http://www.ncbi.nlm.nih.gov/pubmed/11844847.
European Commission. Direttiva 2010/84/UE del Parlamento Europeo e del Consiglio. European Commission. 2010. http://ec.europa.eu/health/files/eudralex/vol-1/dir_2010_84/dir_2010_84_it.pdf.
Drugs and Therapy Perspectives. Practical Issues and Updates: Improved guidelines will optimize efficacy and minimize adverse effects of biosimilar drugs. Drugs Ther Perspect. 2012 [cited 2016 Apr 27];24(8):24–26. http://link.springer.com/article/10.2165/00042310-200824080-00008.
European Medicines Agency. EMA/244682/2013: Medicines under additional monitoring. European Medcines Agency. 2013. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2013/04/WC500142430.pdf.
Calvo B, Zuñiga L. EU’s new pharmacovigilance legislation: considerations for biosimilars. Drug Saf. 2014;37(1):9–18. http://www.ncbi.nlm.nih.gov/pubmed/24190573.
Acknowledgments
The authors declare that no funding has been received for the preparation of this manuscript. SF contributed to writing the entire content of the manuscript. GF and PB contributed to writing part on the content of the manuscript. The authors acknowledge Luigina Mei for assistance in writing this manuscript. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Sara Francescon, Giulia Fornasier, and Paolo Baldo declare that they have no conflict of interest.
Compliance with Ethics Guidelines
This article does not contain any new studies with human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/DBE4F0604F438F43.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Francescon, S., Fornasier, G. & Baldo, P. Biosimilar Oncology Drugs in Europe: Regulatory and Pharmacovigilance Considerations. Oncol Ther 4, 173–182 (2016). https://doi.org/10.1007/s40487-016-0028-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-016-0028-9