
Review of Current Human Genome-Scale Metabolic Models for Brain Cancer and Neurodegenerative Diseases

 ,
,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Literature Search for Manually Curated Brain GEMs
2.2. Inclusion and Exclusion Criteria of Publications
2.3. Metadata Gathering for Determining the Extensiveness of the Manual Curation
2.4. Determining Model Sizes and Common Genes
2.5. Determining the Level of Completeness and Specificity of the Brain GEM
2.6. Evaluation of Objective Function and Validation Used in the Brain GEM
3. Results
3.1. Selected Brain Metabolic Models Could Be Potentially Reused for NDD and Glioma
3.2. Lewis2010 (iNL403)
3.3. Sertbaş2014 (iMS570) from Tunahan Çakır Lab
3.4. Özcan2016 (iMS570g) from Tunahan Çakır Lab
3.5. MartínJiménez2017
3.6. Thiele2020
3.7. Baloni2020
3.8. EcheverriPeña2021 (Neuro-Glia_GEM)
3.9. Lam2021
3.10. Larsson2020 [108]
3.11. Manual Curation Included Tissue-Specific Constraints, Added Reactions, and Compartments
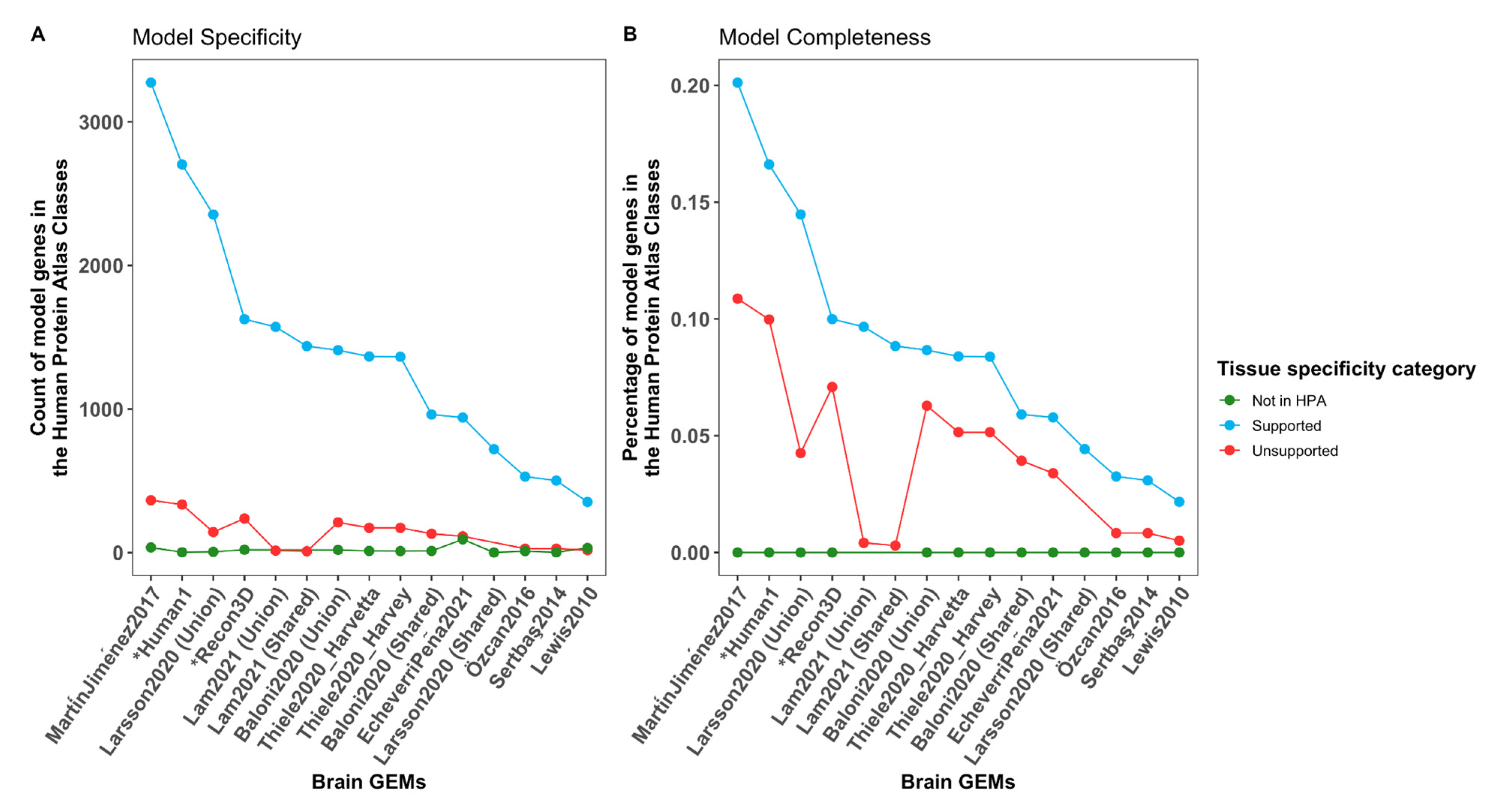
3.12. The Completeness Is Highly Variable between the Models While Having a Similar Specificity
3.13. Glutamine/Glutamate/GABA Exchange Is a Brain-Specific Objective Function for Non-Glioma Models
3.14. GABA and Ornithine Were Included in the Biomass Formulation of a GBM-Specific Biomass Function
3.15. CRISPR-CAS9 Screens, Experimental Fluxes and Simulating Metabolic Dysregulation Are Used as Validation
4. Discussion
4.1. Limitations in the Brain Models Include Non-Standard Reaction Identifiers and the Use of Outdated Model-Building Algorithms
4.2. A High Completeness Is Obtained at the Cost of the Specificity
4.3. Using Standard Identifiers and Confidence Scores Are Required for Model Comparison and Improvement
4.4. The Application of Constraints to the Generic Model Prior to the Context-Specific Model Reconstruction Increases Predictability
4.5. Constraining with Flux Rates Should Be Adjusted to the Generic Model
4.6. Metabolic Tasks of Brain Cell Functions Could Be Employed in Addition to Tailoring the OF
4.7. Bulk Regional Expression Data of the Brain May Serve as an Alternative for Capturing Cellular Heterogeneity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Institute for Health Metrics and Evaluation (IHME). GBDCompareDataVisualization. Seattle, WA: IHME, University of Washington. 2016. Available online: Http://Vizhub.Healthdata.Org/Gbd-Compare (accessed on 28 February 2022).
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Kelley, A.S.; McGarry, K.; Gorges, R.; Skinner, J.S. The Burden of Health Care Costs in the Last 5 Years of Life. Ann. Intern. Med. 2015, 163, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Government’s Missed Opportunity on Research Funding Leaves Dementia Community Disappointed. Available online: https://www.alzheimersresearchuk.org/blog/governments-missed-opportunity-on-research-funding-leaves-dementia-community-disappointed/ (accessed on 28 February 2022).
- World Health Organization. Fact Sheet Cancer. Available online: Https://Www.Who.Int/News-Room/Fact-Sheets/Detail/Cancer (accessed on 1 May 2022).
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6. [Google Scholar]
- Garcia, J.H.; Jain, S.; Aghi, M.K. Metabolic Drivers of Invasion in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 683276. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH Mutation in Glioma: Molecular Mechanisms and Potential Therapeutic Targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Martin-McGill, K.J.; Marson, A.G.; Tudur Smith, C.; Jenkinson, M.D. The Modified Ketogenic Diet in Adults with Glioblastoma: An Evaluation of Feasibility and Deliverability within the National Health Service. Nutr. Cancer 2018, 70, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Z.; Chen, K.; Krepel, S.; Tang, P.; Gong, W.; Zhang, M.; Liang, W.; Trivett, A.; Zhou, M.; Wang, J.M. High Glucose Promotes Human Glioblastoma Cell Growth by Increasing the Expression and Function of Chemoattractant and Growth Factor Receptors. Transl. Oncol. 2019, 12, 1155–1163. [Google Scholar] [CrossRef]
- Yao, P.-S.; Kang, D.-Z.; Lin, R.-Y.; Ye, B.; Wang, W.; Ye, Z.-C. Glutamate/Glutamine Metabolism Coupling between Astrocytes and Glioma Cells: Neuroprotection and Inhibition of Glioma Growth. Biochem. Biophys. Res. Commun. 2014, 450, 295–299. [Google Scholar] [CrossRef]
- Duraj, T.; García-Romero, N.; Carrión-Navarro, J.; Madurga, R.; Ortiz de Mendivil, A.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef]
- Stanke, K.M.; Wilson, C.; Kidambi, S. High Expression of Glycolytic Genes in Clinical Glioblastoma Patients Correlates With Lower Survival. Front. Mol. Biosci. 2021, 8, 752404. [Google Scholar] [CrossRef]
- Qian, Y.; Ding, P.; Xu, J.; Nie, X.; Lu, B. CCL2 Activates AKT Signaling to Promote Glycolysis and Chemoresistance in Glioma Cells. Cell Biol. Int. 2022, 46, 819–828. [Google Scholar] [CrossRef]
- Jin, L.; Zhou, Y. Crucial Role of the Pentose Phosphate Pathway in Malignant Tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [Green Version]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-Based Classification of Glioblastoma Uncovers a Mitochondrial Subtype with Therapeutic Vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef]
- Johung, T.; Monje, M. Neuronal Activity in the Glioma Microenvironment. Curr. Opin. Neurobiol. 2017, 47, 156–161. [Google Scholar] [CrossRef]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.; et al. Altered Metabolic Landscape in IDH-mutant Gliomas Affects Phospholipid, Energy, and Oxidative Stress Pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Ottaviani, L.; Andreoli, R.; Ciociola, T.; Lagrasta, C.A.M.; Tardito, S.; Bussolati, O. Oligodendroglioma Cells Lack Glutamine Synthetase and Are Auxotrophic for Glutamine, but Do Not Depend on Glutamine Anaplerosis for Growth. Int. J. Mol. Sci. 2018, 19, 1099. [Google Scholar] [CrossRef] [Green Version]
- El-Habr, E.A.; Dubois, L.G.; Burel-Vandenbos, F.; Bogeas, A.; Lipecka, J.; Turchi, L.; Lejeune, F.-X.; Coehlo, P.L.C.; Yamaki, T.; Wittmann, B.M.; et al. A Driver Role for GABA Metabolism in Controlling Stem and Proliferative Cell State through GHB Production in Glioma. Acta Neuropathol. 2017, 133, 645–660. [Google Scholar] [CrossRef]
- Blanchart, A.; Fernando, R.; Häring, M.; Assaife-Lopes, N.; Romanov, R.A.; Andäng, M.; Harkany, T.; Ernfors, P. Endogenous GABAA Receptor Activity Suppresses Glioma Growth. Oncogene 2017, 36, 777–786. [Google Scholar] [CrossRef]
- Mulica, P.; Grünewald, A.; Pereira, S.L. Astrocyte-Neuron Metabolic Crosstalk in Neurodegeneration: A Mitochondrial Perspective. Front. Endocrinol. 2021, 12, 668517. [Google Scholar] [CrossRef] [PubMed]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid Accumulation and Oxidation in Glioblastoma Multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef] [Green Version]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.-L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of Tumor Metabolism Reveals Mitochondrial Glucose Oxidation in Genetically Diverse, Human Glioblastomas in the Mouse Brain in Vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Peña-Bautista, C.; Vento, M.; Baquero, M.; Cháfer-Pericás, C. Lipid Peroxidation in Neurodegeneration. Clin. Chim. Acta 2019, 497, 178–188. [Google Scholar] [CrossRef]
- Virtuoso, A.; Giovannoni, R.; De Luca, C.; Gargano, F.; Cerasuolo, M.; Maggio, N.; Lavitrano, M.; Papa, M. The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence. Int. J. Mol. Sci. 2021, 22, 3301. [Google Scholar] [CrossRef]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-Associated Reactive Astrocytes Aid the Evolution of Immunosuppressive Environment in Glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietschy, J.M.; Turley, S.D. Thematic Review Series: Brain Lipids. Cholesterol Metabolism in the Central Nervous System during Early Development and in the Mature Animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Yuan, W.; Chen, T.; Zhou, C.; Liu, C.; Huang, Y.; Han, D.; Huang, Q. HMGCR Positively Regulated the Growth and Migration of Glioblastoma Cells. Gene 2016, 576, 22–27. [Google Scholar] [CrossRef]
- Kambach, D.M.; Halim, A.S.; Cauer, A.G.; Sun, Q.; Tristan, C.A.; Celiku, O.; Kesarwala, A.H.; Shankavaram, U.; Batchelor, E.; Stommel, J.M. Disabled Cell Density Sensing Leads to Dysregulated Cholesterol Synthesis in Glioblastoma. Oncotarget 2017, 8, 14860–14875. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor Initiating Cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Kim, D.K.; Bae, S.-H.; Gwak, H.; Jeon, J.H.; Kim, J.K.; Lee, B.I.; You, H.J.; Shin, D.H.; Kim, Y.-H.; et al. Farnesyl Diphosphate Synthase Is Important for the Maintenance of Glioblastoma Stemness. Exp. Mol. Med. 2018, 50, 137. [Google Scholar] [CrossRef] [Green Version]
- Pirmoradi, L.; Seyfizadeh, N.; Ghavami, S.; Zeki, A.A.; Shojaei, S. Targeting Cholesterol Metabolism in Glioblastoma: A New Therapeutic Approach in Cancer Therapy. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2019, 67, 715–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Zhao, Y.; Gu, Y.; Yang, Y.; Gao, X.; Yuan, Y.; Xiao, L.; Zhang, J.; Sun, C.; Yang, H.; et al. Isocitrate Dehydrogenase 1 Mutation Enhances 24(S)-Hydroxycholesterol Production and Alters Cholesterol Homeostasis in Glioma. Oncogene 2020, 39, 6340–6353. [Google Scholar] [CrossRef]
- Juraszek, B.; Czarnecka-Herok, J.; Nałęcz, K.A. Glioma Cells Survival Depends Both on Fatty Acid Oxidation and on Functional Carnitine Transport by SLC22A5. J. Neurochem. 2021, 156, 642–657. [Google Scholar] [CrossRef]
- Kolar, E.A.; Shi, X.; Clay, E.M.; Moser, A.B.; Lal, B.; Nirujogi, R.S.; Pandey, A.; Bandaru, V.V.R.; Laterra, J.; Pei, Z.; et al. Very Long-Chain Acyl-CoA Synthetase 3 Mediates Onco-Sphingolipid Metabolism in Malignant Glioma. Med. Res. Arch. 2021, 9, 2433. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Shang, E.; Shu, C.; Kim, S.; Mela, A.; Humala, N.; Mahajan, A.; Yang, H.W.; Akman, H.O.; Quinzii, C.M.; et al. Aurora Kinase A Inhibition Reverses the Warburg Effect and Elicits Unique Metabolic Vulnerabilities in Glioblastoma. Nat. Commun. 2021, 12, 5203. [Google Scholar] [CrossRef]
- Menna, G.; Mattogno, P.P.; Donzelli, C.M.; Lisi, L.; Olivi, A.; Della Pepa, G.M. Glioma-Associated Microglia Characterization in the Glioblastoma Microenvironment through a ‘Seed-and Soil’ Approach: A Systematic Review. Brain Sci. 2022, 12, 718. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.-A. The Role of Myelin and Oligodendrocytes in Axonal Energy Metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef]
- Kawashima, T.; Yashiro, M.; Kasashima, H.; Terakawa, Y.; Uda, T.; Nakajo, K.; Umaba, R.; Tanoue, Y.; Tamrakar, S.; Ohata, K. Oligodendrocytes Up-Regulate the Invasive Activity of Glioblastoma Cells via the Angiopoietin-2 Signaling Pathway. Anticancer Res. 2019, 39, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Gomez-Nicola, D. Microglial Dynamics During Human Brain Development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef] [Green Version]
- Lannes, N.; Eppler, E.; Etemad, S.; Yotovski, P.; Filgueira, L. Microglia at Center Stage: A Comprehensive Review about the Versatile and Unique Residential Macrophages of the Central Nervous System. Oncotarget 2017, 8, 114393–114413. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.G.; Lue, L.-F. Immune Phenotypes of Microglia in Human Neurodegenerative Disease: Challenges to Detecting Microglial Polarization in Human Brains. Alzheimers Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 Polarization and Metabolic States. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma Grade Is Associated with the Accumulation and Activity of Cells Bearing M2 Monocyte Markers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune Landscapes Associated with Different Glioblastoma Molecular Subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sielska, M.; Przanowski, P.; Wylot, B.; Gabrusiewicz, K.; Maleszewska, M.; Kijewska, M.; Zawadzka, M.; Kucharska, J.; Vinnakota, K.; Kettenmann, H.; et al. Distinct Roles of CSF Family Cytokines in Macrophage Infiltration and Activation in Glioma Progression and Injury Response. J. Pathol. 2013, 230, 310–321. [Google Scholar] [CrossRef]
- Kumar, V.; Sami, N.; Kashav, T.; Islam, A.; Ahmad, F.; Hassan, M.I. Protein Aggregation and Neurodegenerative Diseases: From Theory to Therapy. Eur. J. Med. Chem. 2016, 124, 1105–1120. [Google Scholar] [CrossRef]
- Grimm, S.; Hoehn, A.; Davies, K.J.; Grune, T. Protein Oxidative Modifications in the Aging Brain: Consequence for the Onset of Neurodegenerative Disease. Free Radic. Res. 2011, 45, 73–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of Pathological Proteins in Neurodegenerative Diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.L.; Pasini, S.; Lambert, W.S.; D’Alessandro, K.B.; Yao, V.; Risner, M.L.; Calkins, D.J. Redistribution of Metabolic Resources through Astrocyte Networks Mitigates Neurodegenerative Stress. Proc. Natl. Acad. Sci. USA 2020, 117, 18810–18821. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.; Maniatis, T. Astrocytes in Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, G.J.; Simkins, T.J.; Emery, B. Neuron-Oligodendrocyte Interactions in the Structure and Integrity of Axons. Front. Cell Dev. Biol. 2021, 9, 653101. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; Khoury, J.E. Microglia in Neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Janda, E.; Boi, L.; Carta, A.R. Microglial Phagocytosis and Its Regulation: A Therapeutic Target in Parkinson’s Disease? Front. Mol. Neurosci. 2018, 11, 144. [Google Scholar] [CrossRef] [Green Version]
- Song, G.J.; Suk, K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wei, Y.-Z.; Wang, G.-Q.; Li, D.-D.; Shi, J.-S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell. Neurosci. 2019, 12, 531. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Liang, J.; Zhou, B. Glucose Metabolic Dysfunction in Neurodegenerative Diseases—New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef]
- de Oliveira, L.G.; de Angelo, Y.S.; Iglesias, A.H.; Peron, J.P.S. Unraveling the Link Between Mitochondrial Dynamics and Neuroinflammation. Front. Immunol. 2021, 12, 624919. [Google Scholar] [CrossRef] [PubMed]
- Bourgognon, J.-M.; Steinert, J.R. The Metabolome Identity: Basis for Discovery of Biomarkers in Neurodegeneration. Neural Regen. Res. 2019, 14, 387–390. [Google Scholar] [CrossRef]
- Pacheco, M.P.; Bintener, T.; Ternes, D.; Kulms, D.; Haan, S.; Letellier, E.; Sauter, T. Identifying and Targeting Cancer-Specific Metabolism with Network-Based Drug Target Prediction. EBioMedicine 2019, 43, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turanli, B.; Zhang, C.; Kim, W.; Benfeitas, R.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Discovery of Therapeutic Agents for Prostate Cancer Using Genome-Scale Metabolic Modeling and Drug Repositioning. EBioMedicine 2019, 42, 386–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated Analyses of Microbiome and Longitudinal Metabolome Data Reveal Microbial-Host Interactions on Sulfur Metabolism in Parkinson’s Disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [Green Version]
- Baldini, F.; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L.; Betsou, F.; Krüger, R.; Thiele, I. Parkinson’s Disease-Associated Alterations of the Gut Microbiome Predict Disease-Relevant Changes in Metabolic Functions. BMC Biol. 2020, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Moolamalla, S.T.R.; Vinod, P.K. Genome-Scale Metabolic Modelling Predicts Biomarkers and Therapeutic Targets for Neuropsychiatric Disorders. Comput. Biol. Med. 2020, 125, 103994. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, A.; Lam, S.; Altay, O.; Li, X.; Yuan, M.; Zhang, C.; Arif, M.; Turkez, H.; Uhlén, M.; Shoaie, S.; et al. Revealing the Molecular Mechanisms of Alzheimer’s Disease Based on Network Analysis. Int. J. Mol. Sci. 2021, 22, 11556. [Google Scholar] [CrossRef] [PubMed]
- Abdik, E.; Çakır, T. Systematic Investigation of Mouse Models of Parkinson’s Disease by Transcriptome Mapping on a Brain-Specific Genome-Scale Metabolic Network. Mol. Omics 2021, 17, 492–502. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Pan, Y.; Nicolazzo, J.A. Impact of Aging, Alzheimer’s Disease and Parkinson’s Disease on the Blood-Brain Barrier Transport of Therapeutics. Adv. Drug Deliv. Rev. 2018, 135, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Rich, L.R.; Harris, W.; Brown, A.M. The Role of Brain Glycogen in Supporting Physiological Function. Front. Neurosci. 2019, 13, 1176. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Vlassis, N.; Pacheco, M.P.; Sauter, T. Fast Reconstruction of Compact Context-Specific Metabolic Network Models. PLoS Comput. Biol. 2014, 10, e1003424. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.P.; John, E.; Kaoma, T.; Heinäniemi, M.; Nicot, N.; Vallar, L.; Bueb, J.-L.; Sinkkonen, L.; Sauter, T. Integrated Metabolic Modelling Reveals Cell-Type Specific Epigenetic Control Points of the Macrophage Metabolic Network. BMC Genomics 2015, 16, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, S.A.; Palsson, B.O. Context-Specific Metabolic Networks Are Consistent with Experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of Genome-Scale Metabolic Models for 126 Human Tissues Using MCADRE. BMC Syst. Biol. 2012, 6, 153. [Google Scholar] [CrossRef] [Green Version]
- Yizhak, K.; Gaude, E.; Le Dévédec, S.; Waldman, Y.Y.; Stein, G.Y.; van de Water, B.; Frezza, C.; Ruppin, E. Phenotype-Based Cell-Specific Metabolic Modeling Reveals Metabolic Liabilities of Cancer. eLife 2014, 3, e03641. [Google Scholar] [CrossRef] [Green Version]
- Zur, H.; Ruppin, E.; Shlomi, T. IMAT: An Integrative Metabolic Analysis Tool. Bioinforma. Oxf. Engl. 2010, 26, 3140–3142. [Google Scholar] [CrossRef]
- Robaina Estévez, S.; Nikoloski, Z. Context-Specific Metabolic Model Extraction Based on Regularized Least Squares Optimization. PLoS ONE 2015, 10, e0131875. [Google Scholar] [CrossRef]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of Anticancer Drugs for Hepatocellular Carcinoma through Personalized Genome-scale Metabolic Modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef]
- Jolivet, R.; Allaman, I.; Pellerin, L.; Magistretti, P.J.; Weber, B. Comment on Recent Modeling Studies of Astrocyte–Neuron Metabolic Interactions. J. Cereb. Blood Flow Metab. 2010, 30, 1982–1986. [Google Scholar] [CrossRef] [Green Version]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and Analysis of Biochemical Constraint-Based Models Using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöstedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An Atlas of the Protein-Coding Genes in the Human, Pig, and Mouse Brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Lewis, N.E.; Schramm, G.; Bordbar, A.; Schellenberger, J.; Andersen, M.P.; Cheng, J.K.; Patel, N.; Yee, A.; Lewis, R.A.; Eils, R.; et al. Formulating Multicellular Models of Metabolism in Tissues: Application to Energy Metabolism in the Human Brain. Nat. Biotechnol. 2010, 28, 1279–1285. [Google Scholar] [CrossRef] [Green Version]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.Ø. Global Reconstruction of the Human Metabolic Network Based on Genomic and Bibliomic Data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef] [Green Version]
- Sertbaş, M.; Ülgen, K.; Çakır, T. Systematic Analysis of Transcription-Level Effects of Neurodegenerative Diseases on Human Brain Metabolism by a Newly Reconstructed Brain-Specific Metabolic Network. FEBS Open Bio 2014, 4, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özcan, E.; Çakır, T. Reconstructed Metabolic Network Models Predict Flux-Level Metabolic Reprogramming in Glioblastoma. Front. Neurosci. 2016, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Martín-Jiménez, C.A.; Salazar-Barreto, D.; Barreto, G.E.; González, J. Genome-Scale Reconstruction of the Human Astrocyte Metabolic Network. Front. Aging Neurosci. 2017, 9, 23. [Google Scholar] [CrossRef]
- Pornputtapong, N.; Nookaew, I.; Nielsen, J. Human Metabolic Atlas: An Online Resource for Human Metabolism. Database 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Sahoo, S.; Heinken, A.; Hertel, J.; Heirendt, L.; Aurich, M.K.; Fleming, R.M. Personalized Whole-body Models Integrate Metabolism, Physiology, and the Gut Microbiome. Mol. Syst. Biol. 2020, 16, e8982. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D Enables a Three-Dimensional View of Gene Variation in Human Metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Baloni, P.; Funk, C.C.; Yan, J.; Yurkovich, J.T.; Kueider-Paisley, A.; Nho, K.; Heinken, A.; Jia, W.; Mahmoudiandehkordi, S.; Louie, G.; et al. Metabolic Network Analysis Reveals Altered Bile Acid Synthesis and Metabolism in Alzheimer’s Disease. Cell Rep. Med. 2020, 1, 100138. [Google Scholar] [CrossRef] [PubMed]
- Echeverri-Peña, O.Y.; Salazar-Barreto, D.A.; Rodríguez-Lopez, A.; González, J.; Alméciga-Díaz, C.J.; Verano-Guevara, C.H.; Barrera, L.A. Use of a Neuron-Glia Genome-Scale Metabolic Reconstruction to Model the Metabolic Consequences of the Arylsulphatase a Deficiency through a Systems Biology Approach. Heliyon 2021, 7, e07671. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Swainston, N.; Fleming, R.M.T.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A Community-Driven Global Reconstruction of Human Metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Shlomi, T.; Cabili, M.N.; Herrgård, M.J.; Palsson, B.Ø.; Ruppin, E. Network-Based Prediction of Human Tissue-Specific Metabolism. Nat. Biotechnol. 2008, 26, 1003–1010. [Google Scholar] [CrossRef]
- Lam, S.; Hartmann, N.; Benfeitas, R.; Zhang, C.; Arif, M.; Turkez, H.; Uhlén, M.; Englert, C.; Knight, R.; Mardinoglu, A. Systems Analysis Reveals Ageing-Related Perturbations in Retinoids and Sex Hormones in Alzheimer’s and Parkinson’s Diseases. Biomedicines 2021, 9, 1310. [Google Scholar] [CrossRef]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A Versatile Toolbox for Metabolic Network Reconstruction and a Case Study on Streptomyces Coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Nookaew, I.; Jacobson, P.; Walley, A.J.; Froguel, P.; Carlsson, L.M.; Uhlen, M.; et al. Integration of Clinical Data with a Genome-Scale Metabolic Model of the Human Adipocyte. Mol. Syst. Biol. 2013, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-Scale Metabolic Modelling of Hepatocytes Reveals Serine Deficiency in Patients with Non-Alcoholic Fatty Liver Disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, I.; Uhlén, M.; Zhang, C.; Mardinoglu, A. Genome-Scale Metabolic Modeling of Glioblastoma Reveals Promising Targets for Drug Development. Front. Genet. 2020, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Zhang, C.; Ji, B.; Mardinoglu, A.; Nielsen, J.; Hua, Q. Logical Transformation of Genome-Scale Metabolic Models for Gene Level Applications and Analysis. Bioinform. Oxf. Engl. 2015, 31, 2324–2331. [Google Scholar] [CrossRef] [Green Version]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 Update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Imanishi, T.; Gojobori, T. H-Invitational Database: Integrated database of human genes. Tanpakushitsu Kakusan Koso 2004, 49, 1937–1943. [Google Scholar]
- Reidegeld, K.A.; Müller, M.; Stephan, C.; Blüggel, M.; Hamacher, M.; Martens, L.; Körting, G.; Chamrad, D.C.; Parkinson, D.; Apweiler, R.; et al. The Power of Cooperative Investigation: Summary and Comparison of the HUPO Brain Proteome Project Pilot Study Results. Proteomics 2006, 6, 4997–5014. [Google Scholar] [CrossRef]
- Çakιr, T.; Alsan, S.; Saybaşιlι, H.; Akιn, A.; Ülgen, K.Ö. Reconstruction and Flux Analysis of Coupling between Metabolic Pathways of Astrocytes and Neurons: Application to Cerebral Hypoxia. Theor. Biol. Med. Model. 2007, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A Draft Map of the Human Proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardlie, K.G.; DeLuca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; Lek, M.; et al. The Genotype-Tissue Expression (GTEx) Pilot Analysis: Multitissue Gene Regulation in Humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; de Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y.; et al. Update of the FANTOM Web Resource: Expansion to Provide Additional Transcriptome Atlases. Nucleic Acids Res. 2019, 47, D752–D758. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, A.P.; Bidkhori, G.; Shoaie, S.; Clarke, E.; Morrin, H.; Hye, A.; Williams, G.; Ballard, C.; Francis, P.; Aarsland, D. Postmortem Cortical Transcriptomics of Lewy Body Dementia Reveal Mitochondrial Dysfunction and Lack of Neuroinflammation. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2020, 28, 75–86. [Google Scholar] [CrossRef]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional Analysis of Multiple Brain Regions in Parkinson’s Disease Supports the Involvement of Specific Protein Processing, Energy Metabolism, and Signaling Pathways, and Suggests Novel Disease Mechanisms. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2005, 137B, 5–16. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, A Potential Therapeutic Target for Early Intervention in Parkinson’s Disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.-U.; Patrick, E.; Komashko, V.; et al. A Molecular Network of the Aging Human Brain Provides Insights into the Pathology and Cognitive Decline of Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational Correction of Copy-Number Effect Improves Specificity of CRISPR-Cas9 Essentiality Screens in Cancer Cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Pirahanchi, Y.; Jessu, R.; Aeddula, N.R. Physiology, Sodium Potassium Pump. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Baumeister, R.; Murphy, C.T.; Heimbucher, T. Metabolic Adaptation to Hypoxia: Do Worms and Cancer Cells Share Common Metabolic Responses to Hypoxic Stress? Cell Death Differ. 2021, 28, 1434–1436. [Google Scholar] [CrossRef]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.P.; Snell, C.; Steers, G.; Turley, H.; Li, J.-L.; Günther, U.L.; et al. Glucose Utilization via Glycogen Phosphorylase Sustains Proliferation and Prevents Premature Senescence in Cancer Cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Bu, G.; Kanekiyo, T.; Zhao, J. Counteracting Alzheimer’s Disease via Somatic TERT Activation. Nat. Aging 2021, 1, 1081–1082. [Google Scholar] [CrossRef]
- Pfau, T.; Pacheco, M.P.; Sauter, T. Towards Improved Genome-Scale Metabolic Network Reconstructions: Unification, Transcript Specificity and Beyond. Brief. Bioinform. 2016, 17, 1060–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, N.; van Heck, R.G.A.; van Dam, J.C.J.; Schaap, P.J.; Saccenti, E.; Suarez-Diez, M. Consistency, Inconsistency, and Ambiguity of Metabolite Names in Biochemical Databases Used for Genome-Scale Metabolic Modelling. Metabolites 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opdam, S.; Richelle, A.; Kellman, B.; Li, S.; Zielinski, D.C.; Lewis, N.E. A Systematic Evaluation of Methods for Tailoring Genome-Scale Metabolic Models. Cell Syst. 2017, 4, 318–329.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for Standardized Genome-Scale Metabolic Model Testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.-E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An Atlas of Human Metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef]
- Pacheco, M.P.; Pfau, T.; Sauter, T. Benchmarking Procedures for High-Throughput Context Specific Reconstruction Algorithms. Front. Physiol. 2016, 6, 410. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Framework and Resource for More than 11,000 Gene-Transcript-Protein-Reaction Associations in Human Metabolism. Proc. Natl. Acad. Sci. USA 2017, 114, E9740–E9749. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Liu, Y. Metabolomics of Glioma. In Cancer Metabolomics: Methods and Applications; Hu, S., Ed.; Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany; Cham, Switzerland, 2021; pp. 261–276. ISBN 978-3-030-51652-9. [Google Scholar]
- Belykh, E.; Shaffer, K.V.; Lin, C.; Byvaltsev, V.A.; Preul, M.C.; Chen, L. Blood-Brain Barrier, Blood-Brain Tumor Barrier, and Fluorescence-Guided Neurosurgical Oncology: Delivering Optical Labels to Brain Tumors. Front. Oncol. 2020, 10, 739. [Google Scholar] [CrossRef]
- Yan, J.; Kuzhiumparambil, U.; Bandodkar, S.; Dale, R.C.; Fu, S. Cerebrospinal Fluid Metabolomics: Detection of Neuroinflammation in Human Central Nervous System Disease. Clin. Transl. Immunol. 2021, 10, e1318. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinski, D.C.; Jamshidi, N.; Corbett, A.J.; Bordbar, A.; Thomas, A.; Palsson, B.O. Systems Biology Analysis of Drivers Underlying Hallmarks of Cancer Cell Metabolism. Sci. Rep. 2017, 7, 41241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, D.A.; Adamson, D.C. Neuronal-Astrocyte Metabolic Interactions: Understanding the Transition into Abnormal Astrocytoma Metabolism. J. Neuropathol. Exp. Neurol. 2011, 70, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; Mo, M.L.; Nakayasu, E.S.; Schrimpe-Rutledge, A.C.; Kim, Y.M.; Metz, T.O.; Jones, M.B.; Frank, B.C.; Smith, R.D.; Peterson, S.N.; et al. Model-Driven Multi-Omic Data Analysis Elucidates Metabolic Immunomodulators of Macrophage Activation. Mol. Syst. Biol. 2012, 8, 558. [Google Scholar] [CrossRef]
- Qiu, R.; Zhong, Y.; Li, Q.; Li, Y.; Fan, H. Metabolic Remodeling in Glioma Immune Microenvironment: Intercellular Interactions Distinct From Peripheral Tumors. Front. Cell Dev. Biol. 2021, 9, 693215. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of Immune Cells in Cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Moustafa, A.A.; Phillips, J.; Kéri, S.; Misiak, B.; Frydecka, D. On the Complexity of Brain Disorders: A Symptom-Based Approach. Front. Comput. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Seidlitz, J.; Nadig, A.; Liu, S.; Bethlehem, R.A.I.; Vértes, P.E.; Morgan, S.E.; Váša, F.; Romero-Garcia, R.; Lalonde, F.M.; Clasen, L.S.; et al. Transcriptomic and Cellular Decoding of Regional Brain Vulnerability to Neurogenetic Disorders. Nat. Commun. 2020, 11, 3358. [Google Scholar] [CrossRef]
- Bordbar, A.; Feist, A.M.; Usaite-Black, R.; Woodcock, J.; Palsson, B.O.; Famili, I. A Multi-Tissue Type Genome-Scale Metabolic Network for Analysis of Whole-Body Systems Physiology. BMC Syst. Biol. 2011, 5, 180. [Google Scholar] [CrossRef] [Green Version]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.-G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An Anatomic Transcriptional Atlas of Human Glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, K.H.B.; Leon, A.J.; Hui, W.; Lee, S.C.-E.; Batruch, I.; Faust, K.; Klekner, A.; Hutóczki, G.; Koritzinsky, M.; Richer, M.; et al. Topographic Mapping of the Glioblastoma Proteome Reveals a Triple-Axis Model of Intra-Tumoral Heterogeneity. Nat. Commun. 2022, 13, 116. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Model | Goal | Model Used as Template | Curation Status | Cell Type | Diseases | Data |
|---|---|---|---|---|---|---|
| Lewis2010 (iNL403) [91] | Building a curated bi-cellular human brain metabolic model to study AD | Recon 1 [92] | Curated | Astrocyte-Neuron | AD | -Human Protein Reference Database [112] -HINV [113] -HUPO brain proteome project [114] -Literature information for transport reactions between compartment -Constraints for neuron cell types. -Microarray data of AD |
| Sertbaş2014 (iMS570) [93] | Identifying biomarker metabolites for six NDD | Çakιr et al., 2007 [115] | Curated | Astrocyte- Neuron | Six NDD | -Microarray of the six NDD -Literature-derived constraints for a healthy brain |
| Özcan2016 (iMS570g) [94] | Metabolic rewiring pathways in three GBM subtypes | Sertbaş2014 | Curated | Astrocyte- Neuron (glutamatergic, GABAergic, cholinergic) | Three GBM subtypes | -Curated growth objective function -Literature-derived constraints for 26 reactions for GBM -Microarray data of the three GBM cell lines |
| MartínJiménez2017 [95] | Building an astrocyte model reconstruction | HMA [96] | Curated | Astrocyte | Hypoxia | -Microarray data of foetal cortical astrocytes -Literature-derived constraints for healthy astrocyte exchange reactions |
| Thiele2020 [97] | Building sex-specific, multi-organ, whole-body model | Recon3D Model [98] | Curated | Whole-brain | -Human Proteome Map [116] - HPA [90] -CSF metabolites from Human Metabolome Database [99] and other resources. -Organ-specific reactions from literature | |
| Baloni2020 [100] | Analysing the effect of bile acid synthesis in AD in different brain regions | Recon3D Model [98] | Semi-curated | Seven brain regions | AD | -RNA-Seq data for brain regions from post-mortem of normal and AD patients -Metabolomics of primary and secondary bile acids from the post-mortem brain samples -BBB reactions from Thiele2020 -Constraints from Lewis2010 -Human Protein Atlas |
| EcheverriPeña2021 (Neuro-Glia_GEM) [101] | Building a bi-cellular neuron-glial model to identify pathways linked to ARSA deficiency | Two tissue AG models from Recon 2 [102] (Glia: MODEL1310110064, neuron: MODEL1310110033) | Semi-curated | Neuron- Glia | Metachromatic leukodystrophy | -Reactions of the sulfatide degradation from the myelin band |
| Lam2021 [104] | Analysing telomeric ageing in AD and PD | iAdipocytes1850 [106] with gprRules from HMR3 [107] | Semi-curated | Whole-brain | AD, BD | -RNA-Seq of healthy brain from HPA [90] & GTEx [117] -CAGE expression of healthy brain samples from FANTOM5 [118] -RNA-Seq of AD and PD brain samples from Rajkumar dataset [119] and Zhang/Zheng dataset [120,121] -Single-cell RNA-Seq of AD and PD brain samples from ROSMAP [122] -Constraints from Baloni2020 [100] |
| Larsson2020 [108] | Predicting non-toxic essential genes for GBM & identifying metabolic pathways for GBM low & high overall survival | 139 AG patient-derived models [109] using HMR2 generic reconstruction [107] | AG | GBM | -RNAseq of TCGA-GBM [110] -Healthy brain GEM from HMA [96] -CRISPR-Cas9 data for GBM [123] |
| Model | Reactions | Consistent Reactions | Metabolites | Genes | Gene Field Format | Number of ENTREZ Genes |
|---|---|---|---|---|---|---|
| Lewis2010 | 1073 | 727 | 987 | 403 | ENTREZ Gene | 403 |
| Sertbaş2014 | 630 | 589 | 523 | 570 | Gene Symbol | 532 |
| Özcan2016 | 659 | 644 | 548 | 569 | ENTREZ Gene | 569 |
| MartínJiménez2017 | 5659 | 4848 | 5007 | 3765 | Ensembl Gene | 3674 |
| Thiele2020_Harvey | 3602 | 3510 | 2201 | 1836 | ENTREZ Transcript | 1548 |
| Thiele2020_Harvetta | 3602 | 3508 | 2203 | 1843 | ENTREZ Transcript | 1551 |
| Baloni2020 * | 5942 (5341–6328) | 5327 (4870–5696) | 3784 (2808–3926) | 1684 (1524–1846) | ENTREZ Transcript | 1409 (1292–1559) |
| EcheverriPeña2021 | 3831 | 3622 | 2473 | 1375 | ENTREZ Transcript | 1148 |
| Lam2021 * | 3283 (3274–3334) | 2774 (2658–2815) | 2122 (2118–2138) | 1523 (1478–1572) | Ensembl Gene | 1516 (1478–1572) |
| Larsson2020 * | 3917 (2226–4877) | 2951 (1382–3276) | 1649 (1178–2086) | 1840 (1103–2034) | Ensembl Gene | 1838 (1102–2031) |
| Model | Objective Function(s) | Rationale for Choosing the OF |
|---|---|---|
| Lewis2010 | ATP demand for both astrocyte and neuron cell: DM_atp(c): atp[c] + h2o[c] => adp[c] + h[c] + pi[c] | Production of the cholinergic neurotransmitter is ATP-dependent. |
| Sertbaş2014 | 1—Maximisation of the sum of glutamate/glutamine/GABA cycles. 2—Setting the value of the sum of the three-cycle fluxes to the optimal solution, then minimising the Euclidean norm of fluxes. | The 1st OF ensures compact coupling of the intercellular exchange between the astrocyte and neuron. The 2nd OF ensures fluxes with minimal utilisation of metabolic enzymes. |
| Özcan2016 | Curated biomass growth reactions: 2.9404 protein + 0.9074 lipid_WM + 0.1091 RNA + 24 ATP => biomass + 24 ADP | Adjusting the contribution of neurons and astrocytes of macromolecules based on their percentage in the white matter, and the macromolecules composition of the white matter. |
| MartínJiménez2017 | (A) ATP production: ADP[m] + 4 H+[c] + Pi[m] => ATP[m] + 3 H+[m] + H2O[m] (B) Glutamate uptake and glutamine release:Glutamate[x] + Glutamine[c] => Glutamate[c] + Glutamine[x] | The 1st OF ensures the consumption of different metabolites for energy production. The 2nd OF resembles the astrocyte role in detoxification of the extracellular glutamate produced by neurons, and secretion of glutamine needed by the neuron. |
| Thiele2020 | The brain model did not have a default OF but rather the model included different OFs for different scenarios: 1—Biomass maintenance 2—Biomass maintenance with no transcription and translation | Biomass maintenance did not include DNA molecules (dgtp[n], dctp[n], datp[n], dttp[n]) as the brain cells do not replicate. The 2nd OF resembles a fasting condition. |
| Baloni2020 | Equal to MartínJiménez2017 | |
| EcheverriPeña2021 | ATP synthesis | Modelling the highly oxidative state of the excited neuron releasing neurotransmitters |
| Lam2021 | ATP synthesis | |
| Larsson2020 | Growth OF of the generic reconstruction HMR2 |
| Model | Strengths | Drawbacks |
|---|---|---|
| Lewis2010 | -Inclusion of a compartment for BBB (EndotheliumAndBlood) with 55 metabolites that can bypass through it (Supplementary File S2, Table S3) -Adding brain cell type-specific reactions from literature (Lewis et al., 2010 [91], Supplementary Table S1) -Comparison with experimental data of cholinergic neurotransmission rate | -The generic reconstruction used as input is outdated and has lots of short-comings |
| Sertbaş2014 | -Constraining with literature-derived constraints. -Comparison with experimental flux ratios for healthy brain cells (Supplementary File S2, Table S6 and S7). | -Using non-standard reaction identifiers in the model -Missing metFormula field that prevents evaluating the stoichiometric consistency |
| Özcan2016 | -Constraining with literature-derived constraints. -Comparison with experimental flux ratios for GBM (Supplementary File S2, Table S9). | -Using non-standard reaction identifiers in the model -Missing metFormula field that prevents evaluating the stoichiometric consistency |
| MartínJiménez2017 | -Constraining with literature-derived constraints (Supplementary File S2, Table S8) -Validation with dysregulated reactions in ischemia (MartínJiménez et al., 2017 [95], Table 4) | -High rate of included genes that are unsupported in brains -The discretization method used for the expression data is not explained -Missing metFormula field that prevents evaluating the stoichiometric consistency |
| Thiele2020 | -Extracting core reactions from literature and other expression data (Supplementary File S2, Table S5) -Defining permeable and impermeable metabolites across the BBB (Supplementary File S2, Table S3) -Defining CSF metabolic composition from different metabolomics data (Supplementary File S2, Table S4) | -Discretization of the Human Proteome Map using a heuristic threshold |
| Baloni2020 | -Updating the list of Thiele2020 for metabolites passing the BBB (Supplementary File S2, Table S3) -Inclusion of constraints from Lewis2010 and OF from MartínJiménez2017 | -Discretization of the expression data using a heuristic threshold -Manual curation on the AG models after model-building with mCADRE. -Gap filling with 389 sink reactions |
| EcheverriPeña2021 | Adding reactions of myelin sheath degradation in oligodendrocyte. | -Individual AG models [102], used for integrating into a neuron-glial model, were built using the outdated MinMax algorithm -Manual curation by adding reactions after integrating the two AG models -Missing metFormula field that prevents evaluating the stoichiometric consistency |
| Lam2021 | -Using an adipocyte GEM with gprRules of the generic HMR3 instead of using the genetic reconstruction itself | |
| Larsson2020 | -Removing essential toxic genes using predefined tasks for a healthy cell. -Validation of the predicted GBM essential genes against CRISPR-Cas9 data. | -AG reconstruction only |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kishk, A.; Pacheco, M.P.; Heurtaux, T.; Sinkkonen, L.; Pang, J.; Fritah, S.; Niclou, S.P.; Sauter, T. Review of Current Human Genome-Scale Metabolic Models for Brain Cancer and Neurodegenerative Diseases. Cells 2022, 11, 2486. https://doi.org/10.3390/cells11162486
Kishk A, Pacheco MP, Heurtaux T, Sinkkonen L, Pang J, Fritah S, Niclou SP, Sauter T. Review of Current Human Genome-Scale Metabolic Models for Brain Cancer and Neurodegenerative Diseases. Cells. 2022; 11(16):2486. https://doi.org/10.3390/cells11162486
Chicago/Turabian StyleKishk, Ali, Maria Pires Pacheco, Tony Heurtaux, Lasse Sinkkonen, Jun Pang, Sabrina Fritah, Simone P. Niclou, and Thomas Sauter. 2022. "Review of Current Human Genome-Scale Metabolic Models for Brain Cancer and Neurodegenerative Diseases" Cells 11, no. 16: 2486. https://doi.org/10.3390/cells11162486