
Metagenomic Insights into Pathogenic Characterization of ST410 Acinetobacter nosocomialis Prevalent in China

Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection
2.2. Metagenomic Sequencing and Taxonomic Classification
2.3. Genome Assembly of the Targeted Pathogen
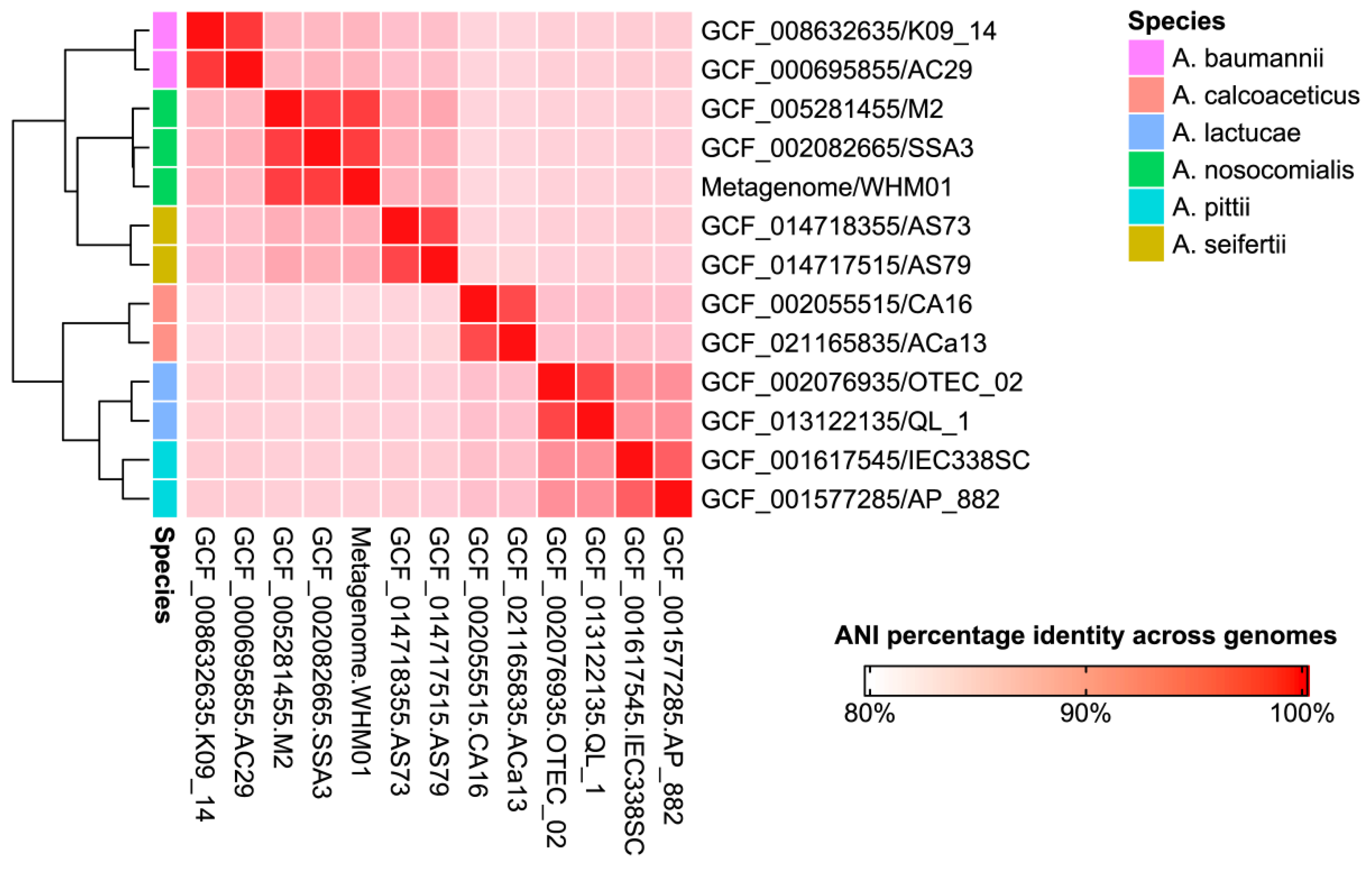
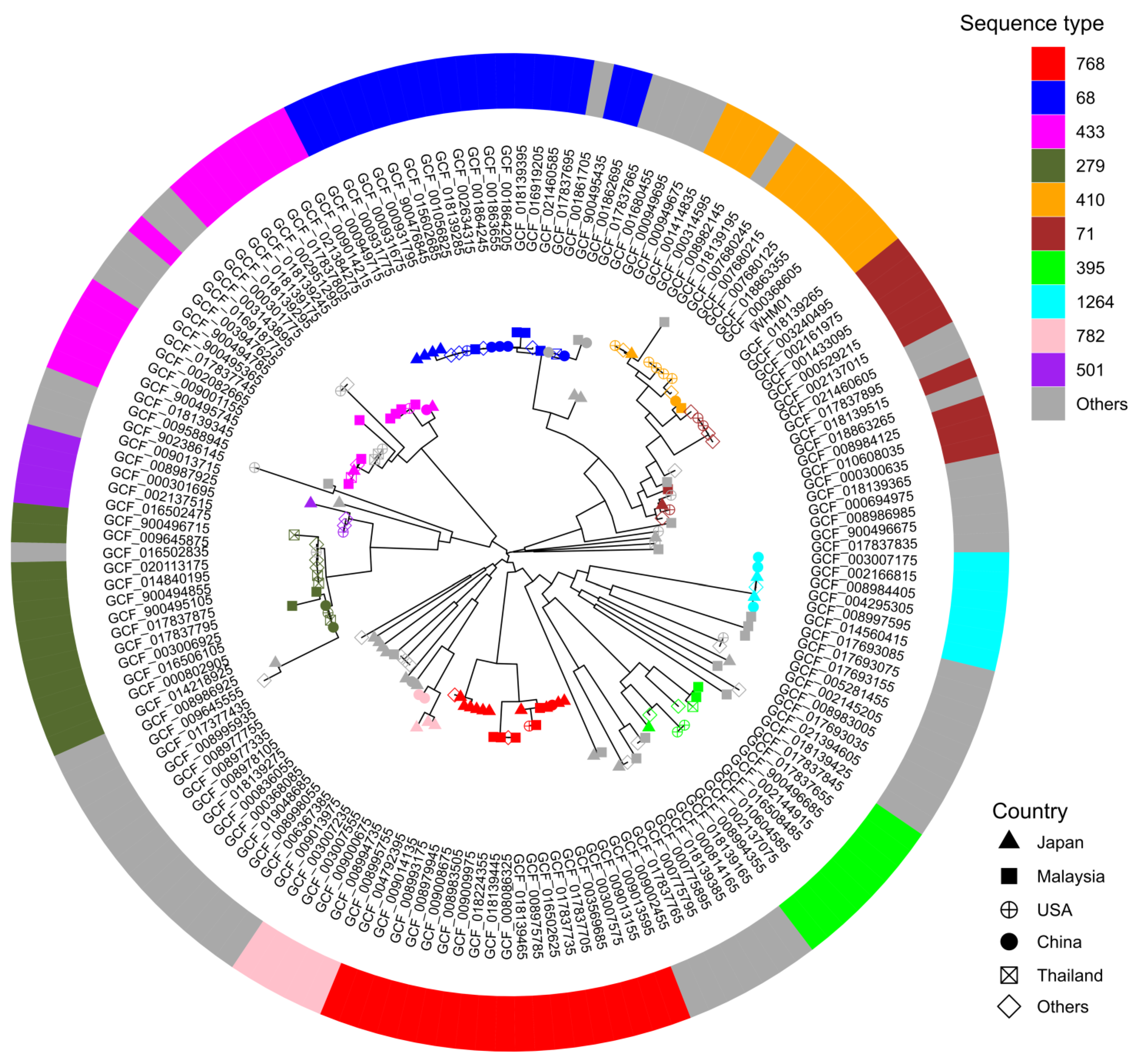
2.4. Analysis of Phylogenome and Pangenome
3. Results
3.1. Pathogenic Risk of A. nosocomialis
3.2. Comparison of Three Assembly Strategies
3.3. Phylogeny of the Genome from the Assembly III
3.4. Virulence-Associated Genes in the Pangenome of A. nosocomialis
4. Discussion
4.1. Clinical Significance of Metagenomic Surveillance on Opportunistic Pathogen
4.2. Genetic Diversity of Virulence Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Visca, P.; Seifert, H.; Towner, K.J. Acinetobacter infection—An emerging threat to human health. IUBMB Life 2011, 63, 1048–1054. [Google Scholar] [CrossRef]
- Cosgaya, C.; Ratia, C.; Marí-Almirall, M.; Rubio, L.; Higgins, P.G.; Seifert, H.; Roca, I.; Vila, J. In vitro and in vivo Virulence Potential of the Emergent Species of the Acinetobacter baumannii (Ab) Group. Front. Microbiol. 2019, 10, 2429. [Google Scholar] [CrossRef] [PubMed]
- Diancourt, L.; Passet, V.; Nemec, A.; Dijkshoorn, L.; Brisse, S. The population structure of Acinetobacter baumannii: Expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS ONE 2010, 5, e10034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Marí-Almirall, M.; Cosgaya, C.; Higgins, P.G.; Van Assche, A.; Telli, M.; Huys, G.; Lievens, B.; Seifert, H.; Dijkshoorn, L.; Roca, I.; et al. MALDI-TOF/MS identification of species from the Acinetobacter baumannii (Ab) group revisited: Inclusion of the novel A. seifertii and A. dijkshoorniae species. Clin. Microbiol. Infect. 2017, 23, 210.e1–210.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.C.; Huang, Y.T.; Tan, C.K.; Kuo, Y.W.; Liao, C.H.; Lee, P.I.; Hsueh, P.R. Acinetobacter baumannii and Acinetobacter genospecies 13TU and 3 bacteraemia: Comparison of clinical features, prognostic factors and outcomes. J. Antimicrob. Chemother. 2011, 66, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Yang, Y.S.; Hsu, W.J.; Chou, Y.C.; Huang, L.S.; Wang, Y.C.; Chiueh, T.S.; Sun, J.R. Emergence of carbapenem-resistant Acinetobacter nosocomialis strain ST410 harbouring plasmid-borne blaOXA-72 gene in Taiwan. Clin. Microbiol. Infect. 2018, 24, 1023–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singkham-in, U.; Chatsuwan, T. Synergism of imipenem with fosfomycin associated with the active cell wall recycling and heteroresistance in Acinetobacter calcoaceticus-baumannii complex. Sci. Rep. 2022, 12, 230. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-J.; Huang, W.-C.; Liao, Y.-C.; Wang, H.-Y.; Lai, J.-F.; Kuo, S.-C.; Lauderdale, T.-L.; Sytwu, H.-K. Molecular Epidemiology of Emerging Carbapenem Resistance in Acinetobacter nosocomialis and Acinetobacter pittii in Taiwan, 2010 to 2014. Antimicrob. Agents Chemother. 2019, 63, e02007–e02018. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho Girão, V.B.; Martins, N.; Cacci, L.C.; Coelho-Souza, T.; Nouér, S.A.; Riley, L.W.; Moreira, B.M. Dissemination of Acinetobacter nosocomialis clone among critically ill patients and the environment. J. Clin. Microbiol. 2013, 51, 2707–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-T.; Chen, H.-Y.; Yang, Y.-S.; Chou, Y.-C.; Chang, T.-Y.; Hsu, W.-J.; Lin, I.C.; Group, A.S.; Sun, J.-R. AdeABC Efflux Pump Controlled by AdeRS Two Component System Conferring Resistance to Tigecycline, Omadacycline and Eravacycline in Clinical Carbapenem Resistant Acinetobacter nosocomialis. Front. Microbiol. 2020, 11, 584789. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-S.; Kuo, S.-C.; Lee, Y.-T.; Yang, Y.-S.; Cheng, S.-S.; Chen, T.-L.; Fung, C.-P. Clinical characteristics and prognostic factors of Acinetobacter nosocomialis bacteraemia in patients with solid tumours. Clin. Microbiol. Infect. 2012, 18, E373–E376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.T.; Kuo, S.C.; Yang, S.P.; Lin, Y.T.; Chiang, D.H.; Tseng, F.C.; Chen, T.L.; Fung, C.P. Bacteremic nosocomial pneumonia caused by Acinetobacter baumannii and Acinetobacter nosocomialis: A single or two distinct clinical entities? Clin. Microbiol. Infect. 2013, 19, 640–645. [Google Scholar] [CrossRef] [Green Version]
- Weber, B.S.; Harding, C.M.; Feldman, M.F. Pathogenic Acinetobacter: From the Cell Surface to Infinity and Beyond. J. Bacteriol. 2015, 198, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.M.; Tracy, E.N.; Carruthers, M.D.; Rather, P.N.; Actis, L.A.; Munson, R.S.; Taylor, R. Acinetobacter baumannii Strain M2 Produces Type IV Pili Which Play a Role in Natural Transformation and Twitching Motility but Not Surface-Associated Motility. Mbio 2013, 4, e00360-13. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.M.; Kinsella, R.L.; Palmer, L.D.; Skaar, E.P.; Feldman, M.F. Medically Relevant Acinetobacter Species Require a Type II Secretion System and Specific Membrane-Associated Chaperones for the Export of Multiple Substrates and Full Virulence. PLoS Pathog. 2016, 12, e1005391. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Scholz, M.; Ward, D.V.; Pasolli, E.; Tolio, T.; Zolfo, M.; Asnicar, F.; Truong, D.T.; Tett, A.; Morrow, A.L.; Segata, N. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods 2016, 13, 435–438. [Google Scholar] [CrossRef]
- Chen, J.; Sun, L.; Liu, X.; Yu, Q.; Qin, K.; Cao, X.; Gu, J. Metagenomic Assessment of the Pathogenic Risk of Microorganisms in Sputum of Postoperative Patients With Pulmonary Infection. Front. Cell. Infect. Microbiol. 2022, 12, 855839. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasolli, E.; Schiffer, L.; Manghi, P.; Renson, A.; Obenchain, V.; Truong, D.T.; Beghini, F.; Malik, F.; Ramos, M.; Dowd, J.B.; et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods 2017, 14, 1023–1024. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab (LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Araya, E.; Muñoz, M.; Rodríguez, C.; Paredes-Sabja, D. FastMLST: A Multi-core Tool for Multilocus Sequence Typing of Draft Genome Assemblies. Bioinform. Biol. Insights 2021, 15, 11779322211059238. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bliss, C.M.; Bennett, J.S.; Bratcher, H.B.; Brehony, C.; Colles, F.M.; Wimalarathna, H.; Harrison, O.B.; Sheppard, S.K.; Cody, A.J.; et al. Ribosomal multilocus sequence typing: Universal characterization of bacteria from domain to strain. Microbiology 2012, 158, 1005–1015. [Google Scholar] [CrossRef]
- Jolley, K.A.; Maiden, M.C.J. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Snippy: Fast Bacterial Variant Calling from NGS Reads. 2015. Available online: https://github.com/tseemann/snippy (accessed on 11 June 2022).
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Lam, T.T.-Y.; Zhu, H.; Guan, Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 2014, 1079, 155–170. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2018, 47, D687–D692. [Google Scholar] [CrossRef]
- Sahl, J.W.; Caporaso, J.G.; Rasko, D.A.; Keim, P. The large-scale blast score ratio (LS-BSR) pipeline: A method to rapidly compare genetic content between bacterial genomes. PeerJ 2014, 2, e332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steimle, A.; Autenrieth, I.B.; Frick, J.-S. Structure and function: Lipid A modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, C.K.; Dalia, T.N.; Klancher, C.A.; Shaevitz, J.W.; Gitai, Z.; Dalia, A.B. Acinetobacter baylyi regulates type IV pilus synthesis by employing two extension motors and a motor protein inhibitor. Nat. Commun. 2021, 12, 3744. [Google Scholar] [CrossRef]
- Doughari, H.J.; Ndakidemi, P.A.; Human, I.S.; Benade, S. The ecology, biology and pathogenesis of Acinetobacter spp.: An overview. Microbes Environ. 2011, 26, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. [Google Scholar] [CrossRef]
- Váradi, L.; Luo, J.L.; Hibbs, D.E.; Perry, J.D.; Anderson, R.J.; Orenga, S.; Groundwater, P.W. Methods for the detection and identification of pathogenic bacteria: Past, present, and future. Chem. Soc. Rev. 2017, 46, 4818–4832. [Google Scholar] [CrossRef]
- Murni, I.K.; Duke, T.; Daley, A.J.; Kinney, S.; Soenarto, Y. True Pathogen or Contamination: Validation of Blood Cultures for the Diagnosis of Nosocomial Infections in a Developing Country. J. Trop. Pediatr. 2017, 64, 389–394. [Google Scholar] [CrossRef]
- Basu, S.; Bose, C.; Ojha, N.; Das, N.; Das, J.; Pal, M.; Khurana, S. Evolution of bacterial and fungal growth media. Bioinformation 2015, 11, 182–184. [Google Scholar] [CrossRef]
- Reidl, J.; Schlör, S.; Kraiss, A.; Schmidt-Brauns, J.; Kemmer, G.; Soleva, E. NADP and NAD utilization in Haemophilus influenzae. Mol. Microbiol. 2000, 35, 1573–1581. [Google Scholar] [CrossRef]
- Opal, S.M. Endotoxins and other sepsis triggers. Contrib. Nephrol. 2010, 167, 14–24. [Google Scholar] [CrossRef]
- Iwashkiw, J.A.; Seper, A.; Weber, B.S.; Scott, N.E.; Vinogradov, E.; Stratilo, C.; Reiz, B.; Cordwell, S.J.; Whittal, R.; Schild, S.; et al. Identification of a general O-linked protein glycosylation system in Acinetobacter baumannii and its role in virulence and biofilm formation. PLoS Pathog. 2012, 8, e1002758. [Google Scholar] [CrossRef]
- Rahn, A.; Beis, K.; Naismith, J.H.; Whitfield, C. A novel outer membrane protein, Wzi, is involved in surface assembly of the Escherichia coli K30 group 1 capsule. J. Bacteriol. 2003, 185, 5882–5890. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Uchida, I.; Terakado, N.; Sasakawa, C.; Yoshikawa, M. Molecular characterization and protein analysis of the cap region, which is essential for encapsulation in Bacillus anthracis. J. Bacteriol. 1989, 171, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Russo, T.A.; Luke, N.R.; Beanan, J.M.; Olson, R.; Sauberan, S.L.; MacDonald, U.; Schultz, L.W.; Umland, T.C.; Campagnari, A.A. The K1 capsular polysaccharide of Acinetobacter baumannii strain 307-0294 is a major virulence factor. Infect. Immun. 2010, 78, 3993–4000. [Google Scholar] [CrossRef] [Green Version]
- Vesel, N.; Blokesch, M. Pilus Production in Acinetobacter baumannii Is Growth Phase Dependent and Essential for Natural Transformation. J. Bacteriol. 2021, 203. [Google Scholar] [CrossRef] [PubMed]
- Tram, G.; Poole, J.; Adams, F.G.; Jennings, M.P.; Eijkelkamp, B.A.; Atack, J.M. The Acinetobacter baumannii Autotransporter Adhesin Ata Recognizes Host Glycans as High-Affinity Receptors. ACS Infect. Dis. 2021, 7, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef]
- He, X.; Lu, F.; Yuan, F.; Jiang, D.; Zhao, P.; Zhu, J.; Cheng, H.; Cao, J.; Lu, G. Biofilm Formation Caused by Clinical Acinetobacter baumannii Isolates Is Associated with Overexpression of the AdeFGH Efflux Pump. Antimicrob. Agents Chemother. 2015, 59, 4817–4825. [Google Scholar] [CrossRef] [Green Version]
- Wilksch, J.; Yang, J.; Clements, A.; Jacinta, L.; Short, K.; Cao, H.; Cavaliere, R.; James, C.; Whitchurch, C.; Schembri, M.; et al. MrkH, a Novel c-di-GMP-Dependent Transcriptional Activator, Controls Klebsiella pneumoniae Biofilm Formation by Regulating Type 3 Fimbriae Expression. PLoS Pathog. 2011, 7, e1002204. [Google Scholar] [CrossRef] [Green Version]
- Gaddy, J.A.; Tomaras, A.P.; Actis, L.A. The Acinetobacter baumannii 19606 OmpA protein plays a role in biofilm formation on abiotic surfaces and in the interaction of this pathogen with eukaryotic cells. Infect. Immun. 2009, 77, 3150–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.H.; Lee, J.S.; Lee, Y.C.; Park, T.I.; Lee, J.C. Acinetobacter baumannii invades epithelial cells and outer membrane protein A mediates interactions with epithelial cells. BMC Microbiol. 2008, 8, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Body Site | No. of Samples | A. nosocomialis | |||
|---|---|---|---|---|---|
| Incidence | Relative Abundance (%) | ||||
| Mean | Max | SD | |||
| Skin | 363 | 18 (4.96%) | 0.0609 | 0.3585 | 0.1120 |
| Oral | 740 | 1 (0.14%) | 0.0002 | 0.0002 | - |
| Stool | 12998 | 7 (0.05%) | 0.1298 | 0.8184 | 0.3050 |
| Nasal cavity | 93 | 0 | - | - | - |
| Vagina | 96 | 0 | - | - | - |
| Assembly I | Assembly II | Assembly III | |
|---|---|---|---|
| No. of contigs | |||
| >500 bp | 226 | 103 | 123 |
| >10,000 bp | 87 | 63 | 62 |
| N50 (bp) | 40,596 | 79,429 | 82,591 |
| NGA50 (bp) | 31,762 | 45,950 | 50,420 |
| Largest (bp) | 173,266 | 172,967 | 173,444 |
| Total (bp) | 3,679,592 | 3,873,672 | 3,892,781 |
| GC (%) | 38.84 | 38.69 | 38.77 |
| Genome fraction (%) a | 86.72 | 87.00 | 87.38 |
| Completeness (%) | 99.86 | 100 | 100 |
| Contamination (%) | 0.14 | 0 | 0 |
| No. of CDSs | 3497 | 3696 | 3704 |
| No. of rRNAs | 2 | 3 | 8 |
| No. of tRNAs | 57 | 58 | 67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jing, L.; Xu, Z.; Zhang, Y.; Li, D.; Song, Y.; Hu, H.; Fang, Y.; Zhu, W. Metagenomic Insights into Pathogenic Characterization of ST410 Acinetobacter nosocomialis Prevalent in China. Pathogens 2022, 11, 838. https://doi.org/10.3390/pathogens11080838
Jing L, Xu Z, Zhang Y, Li D, Song Y, Hu H, Fang Y, Zhu W. Metagenomic Insights into Pathogenic Characterization of ST410 Acinetobacter nosocomialis Prevalent in China. Pathogens. 2022; 11(8):838. https://doi.org/10.3390/pathogens11080838
Chicago/Turabian StyleJing, Liang, Zhuofei Xu, Youping Zhang, Dayong Li, Yaqin Song, Hongjie Hu, Yuan Fang, and Wei Zhu. 2022. "Metagenomic Insights into Pathogenic Characterization of ST410 Acinetobacter nosocomialis Prevalent in China" Pathogens 11, no. 8: 838. https://doi.org/10.3390/pathogens11080838