
Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes
by
 , , , ,
, , , ,
Tal Azagi
1,* ,
,
Ron P. Dirks
2 ,
,
Elena S. Yebra-Pimentel
2,
Peter J. Schaap
3,4,
Jasper J. Koehorst
3,4,
Helen J. Esser
5 and
Hein Sprong
1 1
Centre for Infectious Diseases Research, National Institute for Public Health and the Environment, 3720 BA Bilthoven, The Netherlands
2
Future Genomics Technologies BV, 2333 BE Leiden, The Netherlands
3
Laboratory of Systems and Synthetic Biology, Wageningen University & Research, 6708 PB Wageningen, The Netherlands
4
UNLOCK, Wageningen University, 6708 PB Wageningen, The Netherlands
5
Wildlife Ecology & Conservation Group, Wageningen University, 6708 PB Wageningen, The Netherlands
*
Author to whom correspondence should be addressed.
Microorganisms 2022, 10(6), 1134; https://doi.org/10.3390/microorganisms10061134
Submission received: 13 May 2022
/
Revised: 27 May 2022
/
Accepted: 30 May 2022
/
Published: 31 May 2022
(This article belongs to the Special Issue Ecology, Evolution and Epidemiology of Zoonotic and Vector-Borne Infectious Diseases)
Abstract
:Ca. Neoehrlichia mikurensis is widely prevalent in I. ricinus across Europe and has been associated with human disease. However, diagnostic modalities are limited, and much is still unknown about its biology. Here, we present the first complete Ca. Neoehrlichia mikurensis genomes directly derived from wildlife reservoir host tissues, using both long- and short-read sequencing technologies. This pragmatic approach provides an alternative to obtaining sufficient material from clinical cases, a difficult task for emerging infectious diseases, and to expensive and challenging bacterial isolation and culture methods. Both genomes exhibit a larger chromosome than the currently available Ca. Neoehrlichia mikurensis genomes and expand the ability to find new targets for the development of supportive laboratory diagnostics in the future. Moreover, this method could be utilized for other tick-borne pathogens that are difficult to culture.
1. Introduction
Ixodes ricinus is the most abundant and widespread tick species in Europe [1,2] and transmits multiple pathogens of medical and veterinary concern [3]. Two well-established tick-borne diseases, Lyme borreliosis and tick-borne encephalitis, are frequently reported in Europe, and several studies have indicated a rise in their incidences and spread over the last decades [4,5]. Ixodes ricinus also transmits Ca. Neoehrlichia mikurensis, which has been found all across Europe except for the United Kingdom [6,7,8]. The number of studies describing human infections involving Ca. N. mikurensis is accumulating, but whether these infections result in human disease has not been fully demonstrated [9,10].
One of the major obstacles to investigating how often and under what conditions Ca. N. mikurensis causes an infectious disease in humans, is its unequivocal detection in larger cohorts of patients with noncharacteristic disease symptoms [11,12] or in persons with a (recent) tick bite [13,14,15]. In other words, supportive laboratory diagnostics to detect and identify Ca. N. mikurensis infections are currently limited or reserved to research laboratories. Most importantly, although cultivation of Ca. N. mikurensis has been described in the literature recently [16], it turns out to be quite difficult, even in dedicated laboratories [17,18]. Once one or more Ca. N. mikurensis cultures are generally available, it will become possible to develop more specific and sensitive diagnostic modalities, for example serological tests, which will undoubtedly improve the abilities to detect (endured) infection with Ca. N. mikurensis in clinical practices as well as in epidemiological studies. Recently, genomic information of three Ca. N. mikurensis isolates from Swedish patients became available, giving new insights into its genetic make-up [18].
Since genetic material of emerging tick-borne pathogens from confirmed clinical cases is hard to acquire, our aim was to test whether natural reservoir hosts, a more available source of tissues with high bacteremia, may be utilized as an alternative source for whole genome sequencing. Whole genome sequencing of bacterial and viral pathogens is increasingly used for serotyping and outbreak management [19,20,21]. Previous studies have shown the importance of high-quality DNA as a starting point [22]. Moreover, the use of hybrid assembly strategies, combining long- and short-read sequencing, is recommended for more complete and accurate reference genome assemblies [19,22,23,24,25], especially when repetitive elements are expected. This approach has already been used for other members of the Anaplasmataceae in order to assemble high-resolution whole genomes [26,27]. The long reads provide reliable contigs allowing for repeat regions and complex sequences to be structurally correct, and polishing with accurate short reads corrects errors in the assembly [22,23,28]. Once standardized, an approach in which DNA is extracted following bacterial enrichment, and then sequenced with a hybrid approach, could be used to obtain fully resolved genomes from both host and patient samples.
In this study, we show that two complete and circular Ca. N. mikurensis genomes could be obtained without the need for time- and resource-consuming isolation and culture. These full genomes were directly derived from spleen samples of bank voles (Myodes glareolus) from the Netherlands, using a hybrid assembly approach combining PromethION and Illumina NovaSeq 6000 sequencing. Both genomes exhibit a larger chromosome than the currently available genomes and expand the ability to find new targets for the development of diagnostics in future studies.
2. Materials and Methods
2.1. Sample Collection and Storage
Rodents were collected in various locations in the Netherlands between August and October 2018 (Supplementary Table S1). Rodents were trapped using Heslinga live traps that were filled with hay and baited with a mixture of grains, carrots, and mealworms. Captured rodents were transported to the laboratory facility where they were anesthetized using isoflurane, after which the animals were euthanized by cervical dislocation. Species identification was performed both morphologically and molecularly [29]. For this study, Myodes glareolus were dissected, and spleen samples were taken and stored at −80 °C. All handling procedures were approved by the Animal Experiments Committee of Wageningen University (2017.W-0049.003 and 2017.W-0049.005) and by the Netherlands Ministry of Economic Affairs (FF/75A/2015/014).
2.2. DNA Extraction, Pathogen Detection, and Enrichment
DNA from spleen samples was extracted using the Qiagen DNeasy Blood & Tissue Kit according to the manufacturer’s manual (Qiagen, 2006, Hilden, Germany), and screened for the presence of Ca. N. mikurensis DNA using a qPCR targeting a fragment of the groEL gene (Supplementary Table S2). DNA from two qPCR-positive spleen samples from a male bank vole (samples 18-2804 and 18-2837, Supplementary Table S1) was extracted, this time using the Invitrogen genomic DNA mini kit (Qiagen, Germany) in order to ensure higher genomic DNA yield for NGS. Microbial DNA enrichment was achieved by selective binding and removal of the CpG-methylated host DNA using the NEBNext® Microbiome DNA Enrichment Kit (NEB, Frankfurt am Main, Germany). DNA quality was measured via electrophoresis in Genomic DNA ScreenTape on an Agilent 4200 TapeStation System (Agilent Technologies Netherlands BV, Amstelveen, The Netherlands), and DNA quantity was measured using Qubit dsDNA HS Assay Kit on a Qubit 3.0 Fluorometer (Life Technologies Europe BV, Bleiswijk, The Netherlands).
2.3. Genome Sequencing (Oxford Nanopore Technologies and Illumina)
The DNA from sample 18-2804 was used to prepare a 1D ligation library using the Ligation Sequencing Kit SQK-LSK110 according to the manufacturer’s instructions (Oxford Nanopore Technologies, Oxford, UK). ONT libraries were run on a PromethION flowcell (FLO-PRO002) at Future Genomics Technologies BV (Leiden, The Netherlands) using the following settings: basecall model: high-accuracy; basecaller version: Guppy v4.3.4.
Parallel aliquots of both DNA samples (18-2804 and 18-2837) were used to prepare Illumina libraries using the Nextera DNA Flex Library Prep Kit according to the manufacturer’s instructions (Illumina Inc. San Diego, CA, USA). Library quality was measured via electrophoresis in D1000 ScreenTape on an Agilent 4200 TapeStation System (Agilent Technologies Netherlands BV, Amstelveen, The Netherlands). The genomic paired-end (PE) libraries were sequenced with a read length of 2 × 150 nt using the Illumina NovaSeq 6000 system. Image analysis and basecalling were performed by the Illumina pipeline.
2.4. Genome Assembly and Annotation
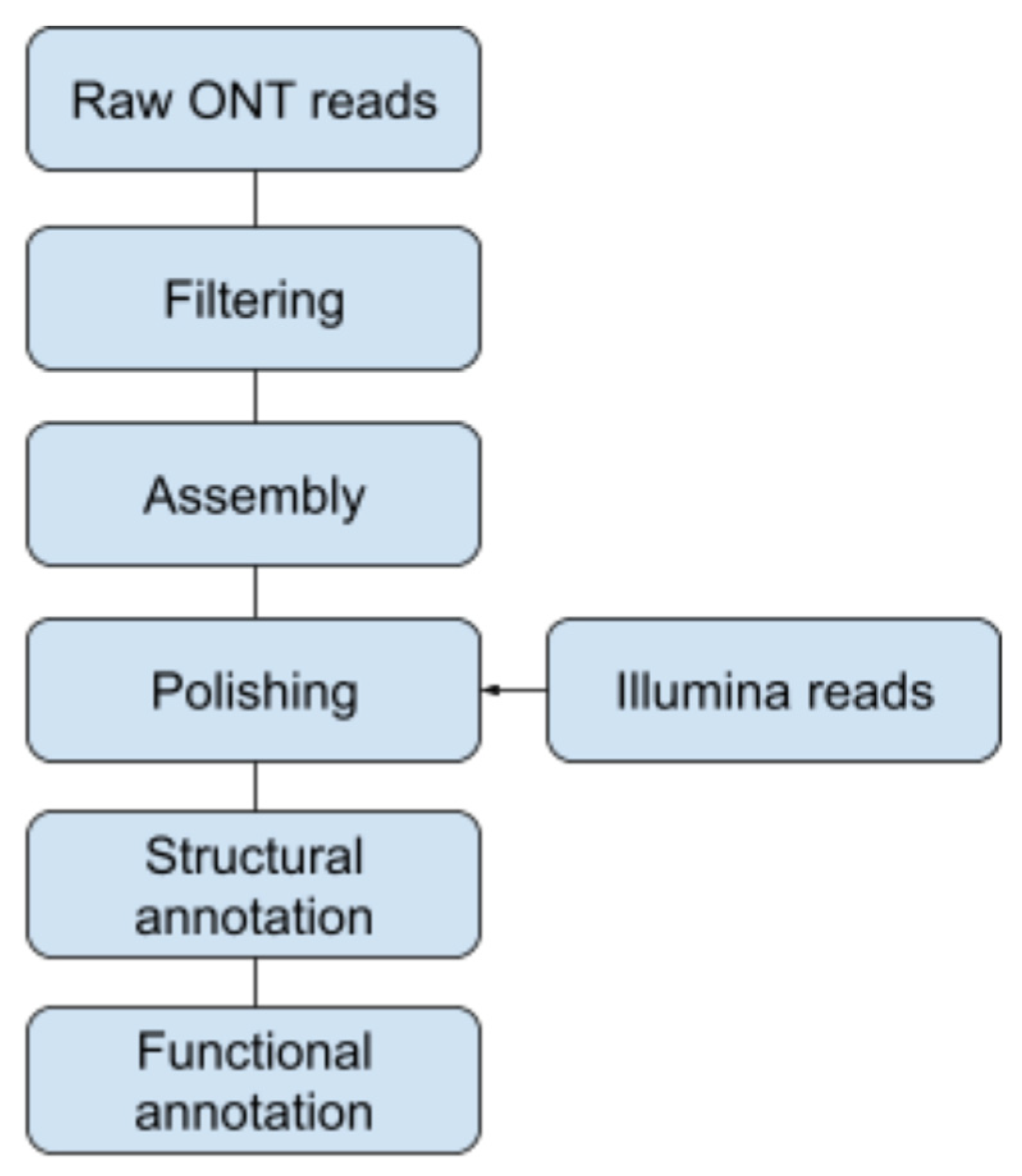
Three reference genomes were used for the removal of host-derived ONT reads: Arvicola amphibious (GCA_903992535.1), Apodemus sylvaticus (GCA_001305905.1), and Myodes glareolus (GCA_004368595.1). Contigs were de novo assembled from the unaligned reads using Flye v2.8.3-b1695 in standard mode and in metagenome mode [30,31]. The “metagenome” contigs were further polished using Medaka v1.4.3 [32]. The filtered reads from the ONT data set were aligned against the Medaka-polished assembly, which were then subsequently used for a de novo assembly using Flye with polishing using Medaka (Figure 1).
Illumina reads of samples 18-2837 and 18-2804 were aligned against the ONT-based consensus sequence of Ca. N. mikurensis (1,236,636 bp; PromethION derived from spleen 18-2804) using minimap2 v 2.17. Pilon vs. 1.23 [33] was then used to polish the ONT-based consensus sequence of Ca. Neoehrlichia mikurensis (18-2804_Ehrlichia_flye_medaka_prokka.fna) independently with the 18-2837 and 18-2804 set of aligned Illumina reads. Prokka v1.14.6 [34] was used to annotate the polished genome sequences. BUSCO v5.2.2 [35] was used for QC of the annotated genome sequences based on the rickettsiales_odb10 lineage dataset. The sorted Illumina reads of 18-2837 and 18-2804 and the sorted nanopore reads of 18-2804 were aligned back against both polished genome sequences to allow visualization of the Bam/Bai files in IGV v2.12.2 [36]. The presence or absence of prophages was determined using the online tool Phaster [37,38]. Furthermore, SAPP [39] was used for the functional annotation of protein coding genes using InterProScan [40] with PFAM [41]. The web version of eggNOG-Mapper v2 [42,43] was used to determine the Clusters of Orthologous Group (COG) categories for protein encoding regions.
2.5. Pangenomic and Comparative Analyses
The Illumina reads of NL07 were mapped to SE20 using minimap2 v2.7 [44], and samtools v1.12 [45] was used to index the bam file and sibeliaZ [46] to generate a maf file. The maf file and bam files were visualized using IGV v2.12.2 and Tablet v1.17.08.17 [47,48], respectively.
The Neoehrlichia pangenome analysis was performed by following the anvi’o pangenomic workflow, and the mcl inflation was set to 2, using anvi’o v7 [49]. For the analyses, the genomes of Ca. N. mikurensis SE24, SE20, SE 26, Ehrlichia chaffeensis Arkansas, Ehrlichia ruminantium Welgevonden, strain Anaplasma phagocytophilum HZ, and Ca. Neoehrlichia lotoris RAC-413 were downloaded from NCBI (accessions numbers are listed in Supplementary Table S3). The Anvi’o genome databases were annotated using the NCBI COG function. A presence–absence Table was used to generate UpSet plots using the R package UpSetR [50] to visualize unique and shared gene clusters at both the intra- and interspecies levels.
2.6. Variant Calling
3. Results
From the 76 M. glareolus captured, 24 spleen samples tested positive for Ca. N. mikurensis DNA in the qPCR analysis (Supplementary Table S1). Four samples with the lowest Ct-values were selected for genomic DNA extraction and microbial enrichment using the NEBNext Microbiome DNA Enrichment Kit (New England BioLabs, Ipswich, MA, USA). The two samples with the highest DNA yield were subjected to genomic sequencing (sample 18-2804 and 18-2837).
3.1. Genomes Generated in This Study
Two complete and circular Ca. N. mikurensis genomes derived from mice spleen samples were assembled in this study. The reference genome derived from PromethION (sample 18-2804) was polished with Illumina data from the same sample, resulting in a circular genome referred to as NL07 (GenBank accession no. CP089285). In addition, the PromethION data derived from sample 18-2804 were polished with Illumina data from sample 18-2837, resulting in a second circular genome referred to as NL06 (GenBank accession no. CP089286). Both assemblies presented a complete genome with high BUSCO scores, which increased from 77.1% to >97% after the short reads were used to polish the long-read assembly and were accurately correct for sequence errors (Table 1, Supplementary Files S1 and S2). No prophages were identified in either genome.
The two genome assemblies generated in this study were compared for strain variations (including both indels and nucleotide substitutions), and in total, 250 variants were found (0.02% difference between genomes). Of these, 153 single nucleotide polymorphisms (SNPs), 34 insertions, 47 deletions, and 16 complex variants were detected. Out of these, two missense variants and three deletions were found in regions pertaining to the P44/Msp2 outer membrane protein (Supplementary Table S4).
As the consensus sequence NL07 presented a higher level of completeness (BUSCO = 99.2%), and both short and long reads were obtained from the same sample, it was used as our reference genome for all downstream analyses. This reference genome (NL07) has 949 coding sequences (CDS) out of 990 genes as well as 3 rRNAs and 37 tRNAs (Table 1). Coding proteins were classified into functional Clusters of Orthologous Group (COG) categories (Supplementary Table S5), and the output was summarized into the number of coding proteins belonging to each COG category (Supplementary Table S6).
3.2. Intraspecies Comparisons
NL07 was compared to three previously published genomes of Ca. N. mikurensis, namely strains SE20, SE24, and SE26, all of which were obtained directly from patient materials in Sweden [18]. Of notable importance, when compared to NL07, the published genomes were on average 124,574 bp smaller. The comparison showed SE26 is the most similar to NL07 (0.02% difference and 165 SNPs), and SE24 is the most distant (0.028% and 257 SNPs) (Table 2). Moreover, when compared to the published strains, NL07 shows mutations that could translate into phenotypic antigenic differences in the coding region of four P44/Msp2 outer membrane proteins (Table 3, Supplementary Tables S7 and S8). In terms of completeness, the BUSCO scores of SE20, SE24, and SE26 are lower than NL07 (93.7%, 94%, 94.3%, and 99.2%, respectively, Supplementary Files S2–S5), which suggests missing conserved genes in the previously published assemblies.
When investigating the 124,574 bp that were absent in the published genomes from patient samples, we found three main expansions that contain 31 genes belonging to 26 known protein domains as well as repeats of the outer membrane protein domain PF01617 (Supplementary Tables S10–S12, Supplementary Figure S1). All but one of the missing genes were most closely related to Ca. Neoehrlichia lotoris. The remaining gene was most similar to a domain participating in biotin metabolism found in Ehrlichia chaffeensis (Supplementary Tables S10 and S11). The Clusters of Orthologous Group (COG) categories assigned to these protein-coding genes are related to various essential processes needed for bacterial survival (Table 4), with the most abundant involved in replication, recombination, and repair (seven protein-coding domains) as well as translation, ribosomal structure, and biogenesis (five protein-coding domains).
The absent genes appear in three main gaps. Upon closer inspection, two of these gaps contain repeats of an outer membrane protein belonging to the Pfam PF01617 domain (Figure 2).
Mapping the Illumina reads of NL07 to the assembly of SE20 shows that many copies of this surface antigen are stacked on top of a site (positioned around 743,000–755,000 bp in SE20) (Supplementary Figure S3). This may be indicative of a collapsed repeat of the surface antigen explaining part of the discrepancy between genome sizes. Moreover, the repeats of the PF01617 domain represent 25 different e-values ranging from 1.8 × 10−6 to 3 × 10−74 that may point to antigenic variation (Supplementary Table S12).
3.3. Pangenome Analysis
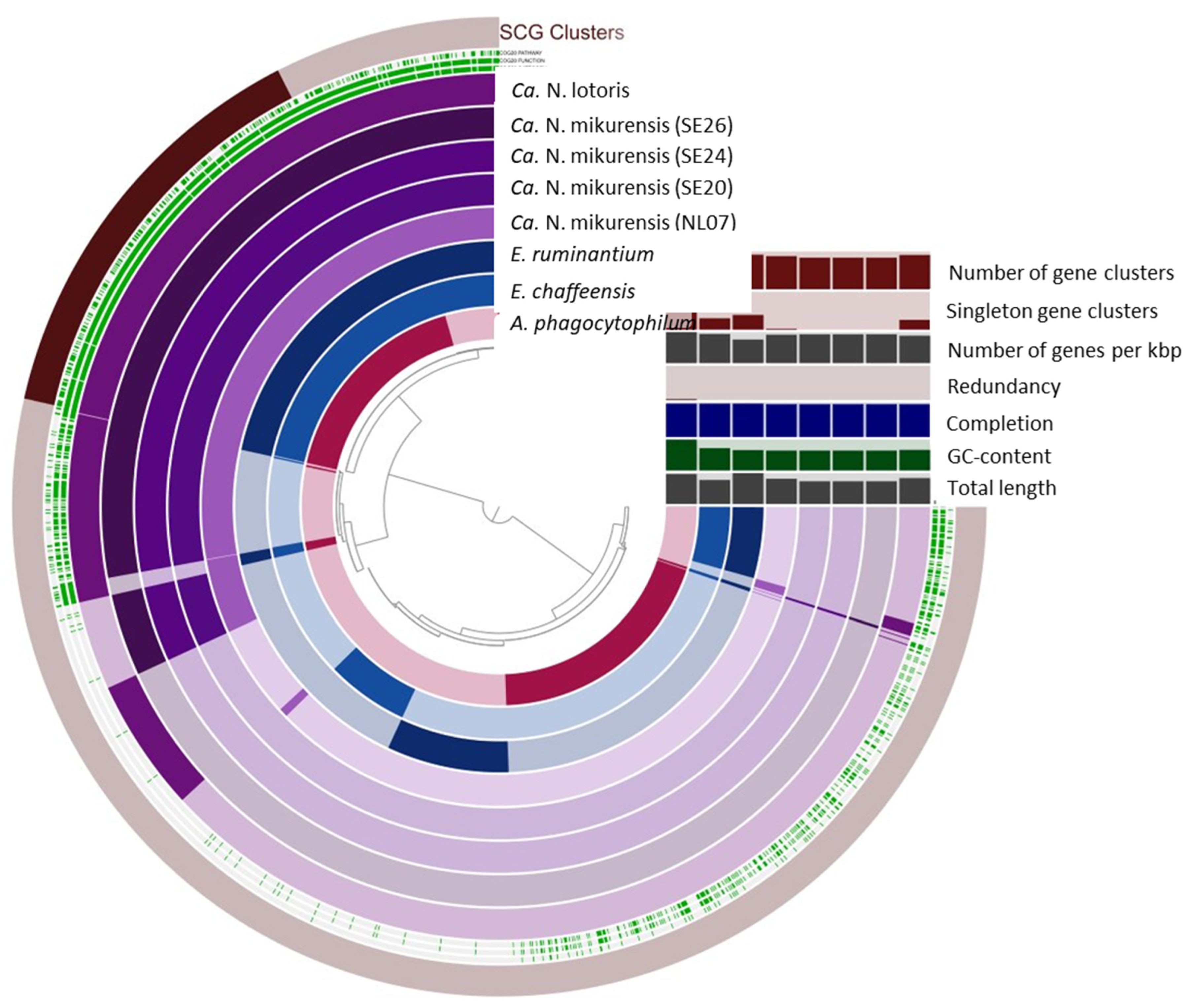
NL07 was compared to select genomes of the Anaplasmataceae family as well as the Ca. N. mikurensis strains from Sweden (Table 5). The GC content of our reference genome (26.85%) is comparable to that of the published strains (26.84%) and close to that of E. ruminantium and Ca. N. lotoris (27.48% and 27.75, respectively) that shares a similar genome size (Table 5, Figure 3). Four hundred and sixty-three gene clusters are present across all genomes (Figure 4). Anaplasma phagocytophilum has the largest genome, and 523 unique gene clusters (Figure 3 and Figure 4). In contrast, NL07 has 13 unique gene clusters and 13 that it only shares with the Ca. N. lotoris genome, the only other genome with which it solely shares gene clusters (Figure 3 and Figure 4). Among the shared clusters are one gene cluster connected to cell motility, two related to cell wall/membrane/envelope biogenesis of which one is an outer membrane protein, one connected to translation, ribosomal structure, and biogenesis, and one connected to inorganic ion transport and metabolism and a TPR-like repeat domain (Supplementary Table S13).
4. Discussion
Two novel and complete Ca. N. mikurensis genomes have been generated in this study using a reproducible approach for high-quality whole genome assembly directly from rodent spleens collected in the wild. These genomes expand on our ability to identify potential targets for the development of reliable diagnostic tools for neoehrlichiosis, which are currently lacking for this and some other tick-borne bacteria.
The genomes presented in this study are approximately 10% larger than the existing Ca. N. mikurensis assemblies recently published, which were derived from clinical samples [18]. The discrepancy in chromosome size might be related to genetic divergence rooted in the provenance of the samples or to the difference in technological platforms and assembly approaches employed.
Although the genes missing in the variants from Sweden are all involved in essential processes, one could argue the gene loss is related to pathogenicity gain as has been shown for other intracellular bacteria [52,53,54,55]. In order to investigate this hypothesis, a larger comparison of host- versus patient-derived genomes must be performed.
The genomes in this study are products of a hybrid assembly approach combining long and short reads, while the genomes from Sweden are based on short reads alone. While short reads are highly accurate at the nucleotide level, they lack the ability to reliably elucidate genome structure [23]. When mapped to the assembly of the Swedish variant SE20, our short-read data of NL07 revealed a large spike containing a repeat of an outer membrane protein domain, which has proven to be highly immunogenic in patients infected with other members of the family Anaplasmataceae [56]. Given that the Swedish variants were assembled based on short reads alone, it is possible that this domain, which appears throughout the genome, collapsed into one locus in said assemblies, explaining part of the discrepancy in genome sizes (Supplementary Figure S2).
The relatively high copy number of this domain could be related to the adaptive immunogenic capabilities of Ca. N. mikurensis [57]. In A. phagocytophilum, this domain has over 113 copies, which has been associated with an increased adaptability to the environment during infection [58], a phenomenon that has been described in the surface protein superfamily (Pfam01617) for A. marginale, E. canis, E. chaffeensis, and E. ruminantium [59]. In A. phagocytophilum, p44/msp2 proteins present strain variability, which could explain why our analyses show SNPs in this domain, between the genomes generated in this study as well as between NL07 and the publicly available Ca. N. mikurensis genomes. Thus, we believe this surface protein family should be studied in depth in order to understand the evolutionary processes involved and how they affect antigenic variation for this potentially emerging pathogen.
The genomes presented in this study provide a foundation for future studies that could explore the antigenic variation of Ca. N. mikurensis. Moreover, we believe that this approach, in which wildlife reservoir host derived tissues are directly used to obtain high-quality whole genomes based on hybrid sequencing, should be employed for other emerging tick-borne pathogens and symbionts.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms10061134/s1, Supplementary Tables S1–S13: 1_qPCR results of spleen samples, 2_Primers, 3_External genomes, 4_SNPs_NL07-NL06, 5_Eggnog_output_NL07, 6_Eggnog_COGs_NL07, 7_SNPs_NL07-SE20, 8_SNPs_NL07-SE24, 9_SNPs_NL07-SE26, 10_Genes NL07 only, 11_Eggnog_ouput_GAP, 12_PF01617 repeats, 13_Anvio output; Supplementary Files S1–S5: BUSCO_NL06_CP089286, BUSCO_NL07_CP089285, BUSCO_SE20_CP054597, BUSCO_SE24_CP066557, BUSCO_SE26_CP060793; Supplementary Figure S1: IGV output showing gaps in SE20; Supplementary Figure S2: tablet visualization showing accumulation of PF01617 repeats.
Author Contributions
Conceptualization, T.A., H.S.; methodology, T.A., R.P.D., E.S.Y.-P., J.J.K., P.J.S. and H.J.E.; material acquisition, T.A. and H.J.E.; formal analysis, T.A., R.P.D., E.S.Y.-P. and J.J.K.; investigation, T.A. and H.S.; writing—original draft preparation, T.A. and H.S.; writing—review and editing, T.A., R.P.D., J.J.K., P.J.S., H.J.E. and H.S.; supervision, H.S.; project administration, T.A. and H.S.; funding acquisition, H.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study is funded by The Netherlands Organization for Health Research and Development (ZonMw, project number 52200-30-07), which has a peer-reviewed grant application, and by the Dutch Ministry of Health, Welfare, and Sports. H.S. is also supported by the NorthTick, European Union, European Regional Development Fund, in the North Sea Region Program. None of the funding organizations had or will have any role in the design or the data analysis and interpretation of the study.
Institutional Review Board Statement
All handling procedures were approved by the Animal Experiments Committee of Wa-geningen University (2017.W-0049.003 and 2017.W-0049.005) and by the Netherlands Ministry of Economic Affairs (FF/75A/2015/014).
Informed Consent Statement
Not applicable.
Data Availability Statement
The genome sequences are present in GenBank under accession numbers CP089285 (NL07) and CP089286 (NL06).
Acknowledgments
We would like to thank Stefanos Siozios, University of Liverpool, for his valuable support in the pangenome analysis performed in this study. P.J.S. and J.J.K. acknowledge the Dutch national funding agency NWO and Wageningen University and Research for their financial contribution to the Unlock initiative (NWO: 184.035.007).
Conflicts of Interest
The authors R.P.D. and E.S.Y.-P., employed by Future Genomics Technologies B.V., have no relevant financial or non-financial interests to disclose. The authors declare no conflict of interest.
References
- Estrada-Peña, A.; Mihalca, A.D.; Petney, T.N. Ticks of Europe and North Africa: A Guide to Species Identification; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Medlock, J.M.; Hansford, K.M.; Bormane, A.; Derdakova, M.; Estrada-Pena, A.; George, J.C.; Golovljova, I.; Jaenson, T.G.; Jensen, J.K.; Jensen, P.M.; et al. Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasites Vectors 2013, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprong, H.; Azagi, T.; Hoornstra, D.; Nijhof, A.M.; Knorr, S.; Baarsma, M.E.; Hovius, J.W. Control of Lyme borreliosis and other Ixodes ricinus-borne diseases. Parasites Vectors 2018, 11, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandekerckhove, O.; De Buck, E.; Van Wijngaerden, E. Lyme disease in Western Europe: An emerging problem? A systematic review. Acta Clin. Belg. 2021, 76, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Kunze, U.; Isw, T.B.E. Tick-borne encephalitis-still on the map: Report of the 18th annual meeting of the international scientific working group on tick-borne encephalitis (ISW-TBE). Ticks Tick-Borne Dis. 2016, 7, 911–914. [Google Scholar] [CrossRef]
- Hansford, K.M.; Fonville, M.; Jahfari, S.; Sprong, H.; Medlock, J.M. Borrelia miyamotoi in host-seeking Ixodes ricinus ticks in England. Epidemiol. Infect. 2015, 143, 1079–1087. [Google Scholar] [CrossRef]
- Hansford, K.M.; Fonville, M.; Gillingham, E.L.; Coipan, E.C.; Pietzsch, M.E.; Krawczyk, A.I.; Vaux, A.G.C.; Cull, B.; Sprong, H.; Medlock, J.M. Ticks and Borrelia in urban and peri-urban green space habitats in a city in southern England. Ticks Tick-Borne Dis. 2017, 8, 353–361. [Google Scholar] [CrossRef]
- Olsthoorn, F.; Sprong, H.; Fonville, M.; Rocchi, M.; Medlock, J.; Gilbert, L.; Ghazoul, J. Occurrence of tick-borne pathogens in questing Ixodes ricinus ticks from Wester Ross, Northwest Scotland. Parasites Vectors 2021, 14, 430. [Google Scholar] [CrossRef]
- Hoper, L.; Skoog, E.; Stenson, M.; Grankvist, A.; Wass, L.; Olsen, B.; Nilsson, K.; Martensson, A.; Soderlind, J.; Sakinis, A.; et al. Vasculitis due to Candidatus Neoehrlichia mikurensis: A Cohort Study of 40 Swedish Patients. Clin. Infect. Dis. 2021, 73, e2372–e2378. [Google Scholar] [CrossRef]
- Azagi, T.; Hoornstra, D.; Kremer, K.; Hovius, J.W.R.; Sprong, H. Evaluation of Disease Causality of Rare Ixodes ricinus-Borne Infections in Europe. Pathogens 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Hoornstra, D.; Harms, M.G.; Gauw, S.A.; Wagemakers, A.; Azagi, T.; Kremer, K.; Sprong, H.; van den Wijngaard, C.C.; Hovius, J.W. Ticking on Pandora’s box: A prospective case-control study into ‘other’ tick-borne diseases. BMC Infect. Dis. 2021, 21, 501. [Google Scholar] [CrossRef]
- Geebelen, L.; Lernout, T.; Tersago, K.; Terryn, S.; Hovius, J.W.; Docters van Leeuwen, A.; Van Gucht, S.; Speybroeck, N.; Sprong, H. No molecular detection of tick-borne pathogens in the blood of patients with erythema migrans in Belgium. Parasites Vectors 2022, 15, 27. [Google Scholar] [CrossRef]
- Jahfari, S.; Hofhuis, A.; Fonville, M.; van der Giessen, J.; van Pelt, W.; Sprong, H. Molecular Detection of Tick-Borne Pathogens in Humans with Tick Bites and Erythema Migrans, in the Netherlands. PLoS Negl. Trop. Dis. 2016, 10, e0005042. [Google Scholar] [CrossRef] [Green Version]
- Markowicz, M.; Schotta, A.M.; Hoss, D.; Kundi, M.; Schray, C.; Stockinger, H.; Stanek, G. Infections with Tickborne Pathogens after Tick Bite, Austria, 2015–2018. Emerg. Infect. Dis. 2021, 27, 1048. [Google Scholar] [CrossRef]
- Azagi, T.; Harms, M.; Swart, A.; Fonville, M.; Hoornstra, D.; Mughini-Gras, L.; Hovius, J.W.; Sprong, H.; van den Wijngaard, C. Self-reported symptoms and health complaints associated with exposure to Ixodes ricinus-borne pathogens. Parasites Vectors 2022, 15, 93. [Google Scholar] [CrossRef]
- Wass, L.; Grankvist, A.; Bell-Sakyi, L.; Bergstrom, M.; Ulfhammer, E.; Lingblom, C.; Wenneras, C. Cultivation of the causative agent of human neoehrlichiosis from clinical isolates identifies vascular endothelium as a target of infection. Emerg. Microbes Infect. 2019, 8, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Raoult, D. Uncultured candidatus neoehrlichia mikurensis. Clin. Infect. Dis. 2014, 59, 1042. [Google Scholar] [CrossRef]
- Grankvist, A.; Jaen-Luchoro, D.; Wass, L.; Sikora, P.; Wenneras, C. Comparative Genomics of Clinical Isolates of the Emerging Tick-Borne Pathogen Neoehrlichia mikurensis. Microorganisms 2021, 9, 1488. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Quainoo, S.; Coolen, J.P.M.; van Hijum, S.; Huynen, M.A.; Melchers, W.J.G.; van Schaik, W.; Wertheim, H.F.L. Whole-Genome Sequencing of Bacterial Pathogens: The Future of Nosocomial Outbreak Analysis. Clin. Microbiol. Rev. 2017, 30, 1015–1063. [Google Scholar] [CrossRef] [Green Version]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B.; et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef]
- Johnson, L.K.; Sahasrabudhe, R.; Gill, J.A.; Roach, J.L.; Froenicke, L.; Brown, C.T.; Whitehead, A. Draft genome assemblies using sequencing reads from Oxford Nanopore Technology and Illumina platforms for four species of North American Fundulus killifish. Gigascience 2020, 9, giaa067. [Google Scholar] [CrossRef] [PubMed]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahara, K.; Suzuki, M.; Hirabayashi, A.; Suda, W.; Hattori, M.; Suzuki, Y.; Okazaki, Y. Long-read metagenomics using PromethION uncovers oral bacteriophages and their interaction with host bacteria. Nat. Commun. 2021, 12, 27. [Google Scholar] [CrossRef]
- Neave, M.J.; Mileto, P.; Joseph, A.; Reid, T.J.; Scott, A.; Williams, D.T.; Keyburn, A.L. Comparative genomic analysis of the first Ehrlichia canis detections in Australia. Ticks Tick-Borne Dis. 2022, 13, 101909. [Google Scholar] [CrossRef]
- Liu, Z.; Peasley, A.M.; Yang, J.; Li, Y.; Guan, G.; Luo, J.; Yin, H.; Brayton, K.A. The Anaplasma ovis genome reveals a high proportion of pseudogenes. BMC Genom. 2019, 20, 69. [Google Scholar] [CrossRef] [Green Version]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, M.; Ali, H.S.; Stieger, N.; Groschup, M.H.; Wolf, R.; Ulrich, R.G. Molecular identification of small mammal species using novel cytochrome B gene-derived degenerated primers. Biochem. Genet. 2012, 50, 440–447. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. metaFlye: Scalable long-read metagenome assembly using repeat graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Medaka: Sequence Correction Provided by ONT Research. Available online: https://github.com/nanoporetech/medaka (accessed on 21 May 2021).
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Koehorst, J.J.; van Dam, J.C.J.; Saccenti, E.; Martins Dos Santos, V.A.P.; Suarez-Diez, M.; Schaap, P.J. SAPP: Functional genome annotation and analysis through a semantic framework using FAIR principles. Bioinformatics 2018, 34, 1401–1403. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Minkin, I.; Medvedev, P. Scalable multiple whole-genome alignment and locally collinear block construction with SibeliaZ. Nat. Commun. 2020, 11, 6327. [Google Scholar] [CrossRef]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet—Next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef] [Green Version]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef]
- Eren, A.M.; Kiefl, E.; Shaiber, A.; Veseli, I.; Miller, S.E.; Schechter, M.S.; Fink, I.; Pan, J.N.; Yousef, M.; Fogarty, E.C.; et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 2021, 6, 3–6. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Snippy: Fast Bacterial Variant Calling from NGS Reads; GitHub, Inc.: San Francisco, CA, USA, 2015. [Google Scholar]
- Georgiades, K.; Merhej, V.; El Karkouri, K.; Raoult, D.; Pontarotti, P. Gene gain and loss events in Rickettsia and Orientia species. Biol. Direct 2011, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Andersson, S.G.; Kurland, C.G. Reductive evolution of resident genomes. Trends Microbiol. 1998, 6, 263–268. [Google Scholar] [CrossRef]
- Fournier, P.E.; El Karkouri, K.; Leroy, Q.; Robert, C.; Giumelli, B.; Renesto, P.; Socolovschi, C.; Parola, P.; Audic, S.; Raoult, D. Analysis of the Rickettsia africae genome reveals that virulence acquisition in Rickettsia species may be explained by genome reduction. BMC Genom. 2009, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Karkouri, K.; Ghigo, E.; Raoult, D.; Fournier, P.E. Genomic evolution and adaptation of arthropod-associated Rickettsia. Sci. Rep. 2022, 12, 3807. [Google Scholar] [CrossRef] [PubMed]
- Rikihisa, Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: Subversive manipulators of host cells. Nat. Rev. Microbiol. 2010, 8, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Van der Woude, M.W.; Baumler, A.J. Phase and antigenic variation in bacteria. Clin. Microbiol. Rev. 2004, 17, 581–611, table of contents. [Google Scholar] [CrossRef] [Green Version]
- Dunning Hotopp, J.C.; Lin, M.; Madupu, R.; Crabtree, J.; Angiuoli, S.V.; Eisen, J.A.; Seshadri, R.; Ren, Q.; Wu, M.; Utterback, T.R.; et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006, 2, e21. [Google Scholar] [CrossRef]
- Noh, S.M.; Brayton, K.A.; Knowles, D.P.; Agnes, J.T.; Dark, M.J.; Brown, W.C.; Baszler, T.V.; Palmer, G.H. Differential expression and sequence conservation of the Anaplasma marginale msp2 gene superfamily outer membrane proteins. Infect. Immun. 2006, 74, 3471–3479. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Overview of the assembly workflow. Raw ONT reads were filtered followed by a draft assembly. The assembly was curated using Illumina reads.
Figure 1.
Overview of the assembly workflow. Raw ONT reads were filtered followed by a draft assembly. The assembly was curated using Illumina reads.

Figure 2.
IGV plot indicating (A) the full assembly of NL07, (B) in blue, the regions of the SE20 assembly that align to NL07 and the gaps that SE20 does not encompass, and (C) in blue, the location of repeats of the outer membrane protein repeats belonging to the PF01617 domain in NL07 and SE20. Note that most repeats are present only in NL07.
Figure 2.
IGV plot indicating (A) the full assembly of NL07, (B) in blue, the regions of the SE20 assembly that align to NL07 and the gaps that SE20 does not encompass, and (C) in blue, the location of repeats of the outer membrane protein repeats belonging to the PF01617 domain in NL07 and SE20. Note that most repeats are present only in NL07.

Figure 3.
Comparative Anvi’o genomic analysis of NL07 and the additional Ca. N. mikurensis, Ca. N. lotoris, E. chaffeensis, E. ruminantium, and A. phagocytophilum genomes included in this study based on the presence/absence of gene clusters. The inner layers represent individual genomes organized by their phylogenetic relationships as indicated by the dendrogram. In the layers, dark colors indicate the presence of a gene group, and light colors indicate its absence. Number of gene clusters, singleton gene clusters, number of genes per kbp, redundancy, completion, GC content, and total length are represented in bar plots. SCG clusters = Single copy gene clusters.
Figure 3.
Comparative Anvi’o genomic analysis of NL07 and the additional Ca. N. mikurensis, Ca. N. lotoris, E. chaffeensis, E. ruminantium, and A. phagocytophilum genomes included in this study based on the presence/absence of gene clusters. The inner layers represent individual genomes organized by their phylogenetic relationships as indicated by the dendrogram. In the layers, dark colors indicate the presence of a gene group, and light colors indicate its absence. Number of gene clusters, singleton gene clusters, number of genes per kbp, redundancy, completion, GC content, and total length are represented in bar plots. SCG clusters = Single copy gene clusters.

Figure 4.
UpSet plot representing the shared and unique gene clusters between NL07 and other members of the Anaplasmataceae family. Black dots indicate the presence of a gene cluster, and connected dots indicate their presence across genomes. The colored horizontal bars represent the amount of gene clusters per genome ranging from 850 to 1018.
Figure 4.
UpSet plot representing the shared and unique gene clusters between NL07 and other members of the Anaplasmataceae family. Black dots indicate the presence of a gene cluster, and connected dots indicate their presence across genomes. The colored horizontal bars represent the amount of gene clusters per genome ranging from 850 to 1018.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Specifications of the unpolished and Illumina polished Ca. N. mikurensis consensus sequences. Note that NL07 was assembled based on PromethION + Illumina data from the same spleen sample (18-2804), while NL06 was assembled based on PromethION data from 18-2804 and Illumina data from 18-2837. All further analyses are based on NL07 only.
Table 1.
Specifications of the unpolished and Illumina polished Ca. N. mikurensis consensus sequences. Note that NL07 was assembled based on PromethION + Illumina data from the same spleen sample (18-2804), while NL06 was assembled based on PromethION data from 18-2804 and Illumina data from 18-2837. All further analyses are based on NL07 only.
| Sample Name | PromethION 18-2804 | PromethION + Illumina 18-2804 (NL07) | PromethION 18-2804 + Illumina 18-2837 (NL06) |
|---|---|---|---|
| Assembly size | 1,236,636 | 1,236,870 | 1,236,136 |
| No. CDS | 1152 | 949 | 958 |
| No. gene | 1193 | 990 | 999 |
| No. rRNA | 3 | 3 | 3 |
| No. tRNA | 37 | 37 | 37 |
| BUSCO score | 77.1% | 99.2% | 97.8% |
Table 2.
Genetic variation between NL07 and SE20 and SE24 and SE26. The Table shows the total amount of variants between NL07 and a given strain (Variant total), the number of multiple nucleotide polymorphisms (Complex), the number of deletions (Deletions), the number of insertions (Insertions), the number of single nucleotide polymorphisms (SNPs), the assembly size (Genome size), and the percentage in difference between NL07 and a given strain in the aligned regions (% difference).
Table 2.
Genetic variation between NL07 and SE20 and SE24 and SE26. The Table shows the total amount of variants between NL07 and a given strain (Variant total), the number of multiple nucleotide polymorphisms (Complex), the number of deletions (Deletions), the number of insertions (Insertions), the number of single nucleotide polymorphisms (SNPs), the assembly size (Genome size), and the percentage in difference between NL07 and a given strain in the aligned regions (% difference).
| Strain | Variant Total | Complex | Deletions | Insertions | SNPs | Genome Size (NL07 = 1,236,870) | % Difference |
|---|---|---|---|---|---|---|---|
| SE20 | 336 | 16 | 30 | 53 | 237 | 1,112,315 | 0.027 |
| SE24 | 349 | 13 | 31 | 48 | 257 | 1,112,301 | 0.028 |
| SE26 | 247 | 21 | 27 | 34 | 165 | 1,112,271 | 0.020 |
Table 3.
P44/Msp2 family outer membrane protein variants between NL07 and Ca. N. mikurensis SE20, SE24, and SE20 assemblies. The effects of the SNPs are presented as synonymous (functionally silent) or nonsynonymous. Nonsynonymous variants, which lead to either a stop codon or a change in protein sequence, are in bold.
Table 3.
P44/Msp2 family outer membrane protein variants between NL07 and Ca. N. mikurensis SE20, SE24, and SE20 assemblies. The effects of the SNPs are presented as synonymous (functionally silent) or nonsynonymous. Nonsynonymous variants, which lead to either a stop codon or a change in protein sequence, are in bold.
| Strain | Type | Nucleotide Position | Effect |
|---|---|---|---|
| SE20 | complex | 917/999 | stop_gained c.917_919delTACinsAAT p.LeuLeu306 |
| snp | 258/903 | synonymous_variant c.258C > T p.Pro86Pro | |
| snp | 792/903 | synonymous_variant c.792T > C p.Pro264Pro | |
| snp | 168/852 | synonymous_variant c.168G > A p.Pro56Pro | |
| snp | 287/852 | missense_variant c.287G > A p.Ser96Asn | |
| snp | 440/816 | missense_variant c.440C > T p.Ala147Val | |
| SE24 | complex | 917/999 | stop_gained c.917_919delTACinsAAT p.LeuLeu306 |
| snp | 792/903 | synonymous_variant c.792T > C p.Pro264Pro | |
| snp | 168/852 | synonymous_variant c.168G > A p.Pro56Pro | |
| snp | 287/852 | missense_variant c.287G > A p.Ser96Asn | |
| snp | 253/936 | missense_variant c.253C > T p.Pro85Ser | |
| SE26 | snp | 552/903 | synonymous_variant c.552A > G p.Gly184Gly |
| snp | 792/903 | synonymous_variant c.792T > C p.Pro264Pro | |
| snp | 168/852 | synonymous_variant c.168G > A p.Pro56Pro | |
| snp | 287/852 | missense_variant c.287G > A p.Ser96Asn | |
| snp | 433/816 | missense_variant c.433G > A p.Glu145Lys |
Table 4.
Clusters of Orthologous Groups assigned to the protein-coding genes found in NL07 and missing in the published Ca. N. mikurensis genomes.
Table 4.
Clusters of Orthologous Groups assigned to the protein-coding genes found in NL07 and missing in the published Ca. N. mikurensis genomes.
| COG Categories | Description | Number of Genes |
|---|---|---|
| L | Replication, recombination, and repair | 7 |
| J | Translation, ribosomal structure, and biogenesis | 5 |
| H | Coenzyme transport and metabolism | 3 |
| C | Energy production and conversion | 2 |
| F | Nucleotide metabolism and transport | 2 |
| M | Cell wall/membrane/envelope biogenesis | 2 |
| P | Inorganic ion transport and metabolism | 2 |
| G | Carbohydrate metabolism and transport | 1 |
| T | Signal transduction mechanisms | 1 |
| U | Intracellular trafficking, secretion, and vesicular transport | 1 |
Table 5.
Summary of analyzed genomes.
| Microorganism | Genome Length | GC Content | Gene Clusters | Singleton Gene Clusters |
|---|---|---|---|---|
| A. phagocytophilum | 1,471,282 | 41.64 | 1018 | 523 |
| E. chaffeensis | 1,176,248 | 30.10 | 886 | 157 |
| E. ruminantium | 1,512,977 | 27.48 | 931 | 203 |
| NL07 | 1,236,870 | 26.85 | 893 | 13 |
| SE20 | 1,112,315 | 26.84 | 850 | 0 |
| SE24 | 1,112,301 | 26.84 | 850 | 0 |
| SE26 | 1,112,271 | 26.84 | 850 | 0 |
| Ca. N. lotoris | 1,268,660 | 27.75 | 923 | 135 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Azagi, T.; Dirks, R.P.; Yebra-Pimentel, E.S.; Schaap, P.J.; Koehorst, J.J.; Esser, H.J.; Sprong, H. Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes. Microorganisms 2022, 10, 1134. https://doi.org/10.3390/microorganisms10061134
AMA Style
Azagi T, Dirks RP, Yebra-Pimentel ES, Schaap PJ, Koehorst JJ, Esser HJ, Sprong H. Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes. Microorganisms. 2022; 10(6):1134. https://doi.org/10.3390/microorganisms10061134
Chicago/Turabian StyleAzagi, Tal, Ron P. Dirks, Elena S. Yebra-Pimentel, Peter J. Schaap, Jasper J. Koehorst, Helen J. Esser, and Hein Sprong. 2022. "Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes" Microorganisms 10, no. 6: 1134. https://doi.org/10.3390/microorganisms10061134
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.