
Abstract
The drug development process is a lengthy and expensive challenge for all involved players. Experience with the COVID-19 pandemic underlines the need for a rapid and effective approval for treatment options. As essential prerequisites for successful drug approval, a combination of high-quality studies and reliable research must be included. To this day, mainly in vivo data are requested and collected for assessing safety and efficacy and are therefore decisive for the pre-clinical evaluation of the respective drug. This review aims to summarize the current state of the art for safety and efficacy studies in pharmaceutical research and industry to address the relevant regulatory challenges and to provide an outlook on implementing more in vitro methods as alternative to animal testing. While the public demand for alternative methods is becoming louder, first examples have meanwhile found acceptance in relevant guidelines, e.g. the OECD guidelines for skin sensitizer. Besides ethically driven developments, also the rather low throughput and relatively high costs of animal experiments are forcing the industry towards the implementation of alternative methods. In this context, the development of orally inhaled drug products is particularly challenging due to the complexity of the lung as biological barrier and route of administration. The replacement of animal experiments with focus on the lungs requires special designed tools to achieve predictive data. New in vitro test systems of increasing complexity are presented in this review. Limits and advantages are discussed to provide some perspective for a future in vitro testing strategy for orally inhaled drug products.
Graphical abstract

Similar content being viewed by others


Introduction
Brief introduction
The directive 2010/63/EU of the European parliament and of the council underlines that the final goal is ‘full replacement of procedures on live animals for scientific and educational purposes’ [1]. But it also states that for safety and efficacy, a ‘regulatory testing’ is necessary from which some requirements can only be fulfilled by animal experiments [1]. An example of a full replacement is the application of in vitro safety assessment of chemicals in the cosmetics industry [2]. The regulation (EC) No. 1223/2009 of the European Parliament and of the Council, which came into force on November 30, 2009, states that animal experiments for testing finished cosmetics are forbidden by law [3, 4]. This regulation underlines the political pressure on developing new alternatives and on the other side the ability of alternatives for predicting human data. For pulmonary drug development, which is nothing else than inhaling chemicals with an intended safe use and a specific efficacy, several in vitro methods are available but animal experiments are still regulatory required. Within this review, we will summarize and discuss the aspects of chemical registration and drug approval in the context of a potential animal testing free drug development.
The price we pay
Animal experiments are quite expensive, in some cases ethically unacceptable, and they often produce results that are not valid enough to predict potential safety issues in humans [5, 6]. In 2011, 1.3 billion € was spent on animal experiments for the safety assessment of chemicals in total, whereby the acute inhalation toxicity is calculated with 13.85 million € and sub-chronic toxicity studies with 61.95 million € [7]. In the first report for the ‘EU statistical data on the use of animals for scientific purposes’ in 1994, 11,790,485 animal experiments were conducted in the European Union [8]. The total number of animals was decreased to 9,390,000 in 2017, which is communicated in the newest report from the European Commission in 2020 [9].
Next to these considerable costs, the predictivity of animal experiments is called into question by numerous studies. Only 71% of two-species studies can predict the human toxicity studies [10], resulting in a 89% failure rate of new drugs after human clinical trials [5]. Reasons for this low predictivity are physiological differences between the species that lead to, among other things, diverse metabolism functions, varied microbiome constitutions, altered gene expression profiles and differently expressed disease phenotypes [10].
In response to these limitations of animal experiments, the demand for suitable in vitro methods is increasing. Goh et al. calculated all in vitro assays performed in the main three fields genotoxicity, safety pharmacology and ADME of pre-clinical studies from three pharma companies and three contract research organizations (CRO) in the years 1980 to 2013 with the result that the percentage of performed in vitro assays in the pharma industry has increased by a remarkable 20% since 2012 [11]. The actual market of in vitro toxicology testing was estimated at $ 22.7 billion in 2020 and extrapolations demonstrate that the market size of the in vitro toxicology testing will be increased up to 11.4% in 2028 [12]. This increasing number of in vitro assays used for scientific purposes can be related to a slight reduction in animal testing from 1990 until 2017 even if there are strict regulatory requirements.
Regulatory requirements for safety assessment of chemicals and pharmaceuticals
The European Union’s REACH regulation ((EC) 1907/2006) legally regulates the market and safety of chemicals since 2007. According to the slogan ‘no data, no market’, chemical manufacturers must provide all necessary data for a detailed safety evaluation for the product that they want to release to the market [13]. The best way for a uniform evaluation of the product is to adhere to certain guidelines. Relating thereto, the OECD, an international association with 36 member states aiming for the reduction of economic, political and environmental complications through standardized procedures, is responsible for the development of such guidelines [14]. The internationally accepted OECD guidelines include the (bio)safety assessment of chemicals and the protection of the environment and human health. This collection of guidelines for the testing of chemicals is divided into 5 sections: (1) physical–chemical properties; (2) effects on biotic systems; (3) environmental fate and behaviour; (4) health effects and (5) other test guidelines [15]. In the 4th section of guidelines, 80 OECD regulations describe suitable methods for identifying potential health risks of the substances under test. For example, the (acute) toxicity, genotoxicity, neurotoxicity, sensitization and corrosion effects of the test substance are addressed in in vitro and in vivo studies depending on the organ affected [16]. After the aforementioned health risks for human and environment can be assessed, the manufacturer has to apply to the ECHA (European Chemical Agency) for registration, depending on the production volume per year. If approved, the chemical can be placed on the market [17].
In contrast to chemicals, drugs have to demonstrate safety and efficacy for human application. As soon as a potent drug candidate has been identified, several development phases must be passed through. Often, the development process starts with a chemical optimization of the active compound, which serves to improve the physiochemical and biological characteristics to achieve an increased efficacy (lead compound optimization) [18]. In the pre-clinical phase, the targeted compound is tested mainly in short- and long-term animal studies for toxicology and disease-modifying effects [19].
In principle, the pre-clinical testing for orally inhaled drug products is the same as for other routes of applications as parenteral or oral. The manufacturer must provide data which include especially in vivo repeat-dose toxicology (up to 9 months) studies. These studies are performed to exclude toxic effects directly in the lung tissue and avoid systemic toxicity. This will include respiratory tract malfunctions, e.g. larynx and nasal cavity irritancy. As examples, common drugs for inhalation, e.g. salbutamol, fluticasone or tobramycin, were tested in this way [20, 21]. All these studies are performed in vivo, it is recommended to use one rodent and one non-rodent species to evaluate the safety of the drug formulation [21]. A first goal of this testing is the identification of the no-observed-adverse-effect levels (NOAELs) which specify the initial dose for starting the clinical phase I [19, 22].
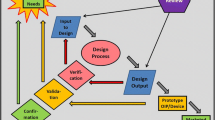
If there are no safety concerns after the pre-clinical phase, the developing company can submit an approval for investigatory new drug (IND) to the US Food and Drug Administration (FDA) or clinical trial application (CTA) by the European Medicines Agency (EMA) [23]. After authorisation has been obtained from the IND or CTA, respectively, the clinical trials start. With an increasing number of participants in the clinical phase, the safety and efficacy of the IND are evaluated in humans until the data situation allows for the preparation of a new drug application (NDA) which is then sent to the FDA or EMA. An independent advisory committee of the agency will discuss the results of the NDA and decide if the drug promises good treatment options for the patients [22, 24]. During the final drug approval phase in Europe, the different national committees can sometimes also be involved in the ultimate approval decision [24]. Occasionally, the regular drug approval process can be accelerated due to an emergency, like during the COVID-19 pandemic, where the BioNTech/Pfizer vaccine received approval after 10 months, compared to an average of 15 years or longer in the regular approval process [25]. Moreover, drug approval processes can be changed when it comes to the approval of orphan drugs including a reduced clinical phase II in 4 years compared to 6 years for standard approvals [24, 26, 27]. Figure 1 summarizes the drug development process which must be passed for a final FDA approval.

copyright © 2020 FDA homepage, https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/new-drug-development-and-review-process, accessed 01/03/2021
Overview of the drug development and approval process of new drugs by the FDA. An investigational new drug (IND) undergoes during their development various phases of pre-clinical research and clinical studies. After a successful synthesis and purification, the IND will be tested in the pre-clinical phase for their efficacy and safety by inter alia short- and long-term animal studies followed by an official IND submission. Passing the three phases of clinical studies, the advisory committees decide for an approval of the drug product [22]. Reprinted from: ‘New Drug Development and Review Process’,
As mentioned above a key element during the drug development process is the IND application, which is crucial for the start of clinical studies in humans. It stands to reason that potential safety issues of a new drug should be clarified in advance. This is done in IND enabling studies which cover the pre-clinical safety assessment by a complete description of the pharmacodynamics and kinetics (ADME properties), safety pharmacology, reproductive and developmental toxicity, and genotoxicity studies [28]. The FDA provides various forms which must be submitted for IND application. One of the most important forms is the FDA 1571 Title 21, Code of Federal Regulations (CFR) Part 312 including a detailed description of the data situation in the pre-clinical evaluation [28,29,30,31]. To ensure FDA acceptance of the data integrated in an IND application, internationally accepted good laboratory and manufacturing practice (GLP/GMP) and quality guidelines from the ‘Organisation for Economic Co-operation and Development’ (OECD) and the ‘International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use’ (ICH) should be met [32,33,34].
The ICH, a consortium of key players from regulatory authorities and pharmaceutical industries, has established standards on quality, safety and efficacy, and also provides the multidisciplinary guidelines, all of which help to unify the international drug development process [35]. These guidelines ensure that adequate data quality can be guaranteed, which makes a successful drug approval more likely. The ICH guidance document M3 (R2), for example, describes in detail the required data for the pre-clinical evaluation of pharmaceuticals to support subsequent clinical trials [36, 37]. Safety assessments do not only play an essential role during drug approval, they are also important for the registration and market authorization of chemicals and, consequently, their commercial distribution and industrial processing [38].
In order to adequately comply with the law and, thus, ensure safety for humans and the environment, animal experiments are still required [39] and are essential from regulatory perspective (e.g. ECHA) as no adequate and valid alternatives are available [40].
Changing the perspective: replacing animal experiments by predicting human data
It is an obvious strategy that animal experiments might be replaced most efficiently by methods leading to the same results as animal experiments do. This concept will transfer all disadvantages such as low predictivity of human data as well. This low predictivity is mainly localized in efficacy and not safety-related questions. From this perspective and with focus on pulmonary drug development, a particular understanding of relevant lung diseases is necessary before developing adequate in vitro tools.
According to the World Health Organization (WHO), chronic obstructive pulmonary disease and lower respiratory infections occupy the 3rd and 4th places, respectively, of the most common causes of death worldwide [41]. The lethality of patients suffering from lung diseases will rise in the future due to the COVID-19 pandemic [42]. The most relevant lung disorders are divided into four main categories: (1) acute lung diseases, e.g. pneumonia and influenza; (2) chronic inflammatory diseases, e.g. chronic obstructive pulmonary disease (COPD) and asthma; (3) occupational lung disorders, such as various forms of lung fibrosis; and (4) parenchymal lung diseases, mostly related to immune disorders [43].
Chronic lung complications are a cause of underlying diseases mostly because of previous acute lung failures such as infections [44]. Chronic lung conditions such as asthma and COPD are mainly triggered by allergies and an increased inflammatory response due to air contaminations, e.g. cigarette smoke and noxious gases [45, 46]. Acute lung diseases such as pneumonia are caused by lower respiratory tract infections of bacteria or viruses [47], resulting mostly in acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) depending on the symptom severity [48,49,50]. This diversity of lung diseases leads to an enormous range of treatment options. For example, in the ‘Guidelines for the management of adult lower respiratory tract infections’, the European Respiratory Society (ERS) has listed various antibiotic treatment options, e.g. amoxicillin or tetracyclines, depending on the degree and type of infection, and recommended that vaccination is suitable for risk prevention [51]. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) compiled a list with medical formulations and typical doses for the pharmacological treatment of COPD. Next to inhalable corticosteroids, anticholinergics, beta2-agonists and various combinations can be applied via metered dose inhaler (MDI) or dry powder inhaler (DPI) to COPD patients [52, 53].
Apart from these classic pharmacological interventions, innovative therapies, especially for the cure of ARDS due to the spread of COVID-19, are on their way for regulatory approval. At the end of 2019, Silva et al. discussed that personalized medicine can offer targeted therapies including coordinated treatments for the individual patient’s biochemical and physiological reconstitution during ARDS [54]. In addition, cell therapies with (embryonic) stem cells promise in vivo a reconstitution of damaged lung epithelial cells during ALI/ARDS, whereas innovative gene therapies can influence protein expression responsible for the regulation of inflammatory signalling pathways [55]. A lot of active pharmaceutical ingredients (API) with potential ARDS treatment options are currently in the pre-clinical phase of drug development. Examples of such APIs are common anti-coagulants with effects against vascular dysfunction, immunomodulatory pleiotropic and pathway-specific consequences (e.g. elafin and anti-INF-γ therapies), and anti-viral agents such as remdesivir and favipiravir which have been heavily promoted in their development during the COVID-19 pandemic in 2020 [56]. A further example for an innovative ARDS treatment is the hormone therapy based on PEG-adrenomedullin submitted as an orphan drug approval by Bayer AG Pharmaceuticals with indication for ARDS in clinical phase II [57, 58]. Next to the local treatment of lung diseases, the inhalative application route, e.g. for insulin therapy or against hypertension, has various advantages, as a faster treatment, higher efficacy and a reduced systemic side effect than other ways of administration [59]. These drug developments and approvals are essential for new therapeutic options to treat the above-mentioned lung diseases. Nevertheless, only a few drug candidates pass the long and financially risky development process to eventually achieve official approval [60]. The reasons for the high failure rate and how in vitro models can be advantageously involved in the drug development process, also considering regulatory aspects, are described in the following section.
Validation is the challenge to face
Significant progress was achieved by replacing animal experiments with focus on skin data, such as the skin sensitization test (No. 442 C-E) and the in vitro skin irritation test (No. 439), and also many in vitro genotoxicity tests, as the mammalian cell micronucleus test (No. 487). However, no in vitro method for the inhalative safety assessment is yet recommended by the OECD. Consequently, the required data must still be generated by animal experiments. The official guidelines for the applied in vivo methods are No. 403, 433, and 436, reporting the acute toxicity, and the guidelines no. 412 and 413, analysing the sub-acute and sub-chronical effects to the lung after substance exposure (Table 1).
On December 12, 2013, the European Union Reference Laboratory for Alternatives to Animal Testing (EURL ECVAM) recommended the Direct Peptide Reactivity Assay (DPRA) testing to evaluate potential skin sensitizers [61]. The methods for in vitro evaluation of skin sensitizer were expanded to the cell-based assays KeratinoSens™, LuSens, h-CLAT, (m)MUSST showing a better predictivity to human data [62]. Urbisch et al. show that in silico methods as the QSAR (quantitative structure–activity relationship) Toolbox and TIMES (tissue metabolism simulator) were more reliable to identify skin sensitizer in comparison to the in vivo LLNA (local lymph node assay) [63]. In 2018, the ECHA recommended the in vitro skin sensitizing methods within the safety assessment to meet the REACH regulation according to the OECD guidelines 442 C-E [64]. In addition, other in vitro test guidelines are officially accepted, e.g. test no. 498: in vitro phototoxicity assay, test no. 490: in vitro mammalian cell gene mutation assay, and test no. 473: in vitro mammalian chromosomal aberration test [65]. A complete list of all validated in vitro test methods is given by the EURL ECVAM on their web page [66]. Although lot of in vitro assays, for examining the skin sensitizer, eye corrosion and gene mutation, were successfully established in guidelines, no officially OECD test guidelines are available for an inhalation toxicity assessment of chemicals. To guide developers of in vitro methods to a potential validation and approval of their in vitro assays, the OECD published the ‘Guidance Document on Good In Vitro Method Practices (GIVIMP)’ in 2018. Based on GMP/GLP system, all important points during the establishment and application of an in vitro method are considered in this GIVIMP guideline. Starting with the qualification of the suppliers of the lung cell cultures (e.g. ATCC, DSMZ), to the necessary quality assurance (QA) and quality control (QC) during validation and application of the method, is described in detail. Further rules, such as the equipment of the facilities, as laminar flow benches, needed material (e.g. cell culture vessels, pipettes) and reagents (e.g. cell culture media, chemicals, supplements), must also be audited and must comply with the GMP/GLP system. A suitable reference/control representing the applicability of the method must be selected within the assay, for example, in a cell viability assay, the unexposed control with buffer and a negative control with a cell destroying detergent. The results must be sufficiently documented and archived so that anyone can check the results for transparency, reproducibility, robustness and accuracy at any time [67]. There is currently no officially recognized in vitro method that fulfils the requirements just mentioned. But many systems are promising, are well-established and are in the starting blocks for validation. They differ in their complexity from simple monolayer 2D cultures to mixed cultures (co-cultures), 3D cultures and overly complex co-cultures with more than three cell types. The costs of these test systems increase but the physiological relevance grows as well. But none of the test systems is validated under permitted guidelines recommended from the OECD or ICH [68]. A complete validation of an applicable in vitro method implies a successful transfer from research to industry [68, 69]. The coordination of this validation process is carried out by the US Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) [70] and, for the European Union, by the EURL ECVAM [71]. In the ECVAM status report from 2019, next to various in vitro assays, e.g. CATMoS (in silico models of acute oral systemic toxicity), in vitro cytotoxicity test 3T3 Neutral Red Uptake and the MELN® human estrogen receptor transcriptional activation assay, only two alternatives for respiratory in vitro alternatives are submitted for validation: (1) the in vitro system ALIsens, which should replace the respiratory local lymph node (LLNA) assay, and (2) undergoing validation at the time of publication, the EpiAirway™ system, a reconstructed human lung epithelium for detecting acute inhalation toxicity studies [72].
The potential power of in vitro assays for chemical and pharmaceutical industry
Alternatives to animal experiment will dominate the future for ethical and economic reasons. The overall advantage is easily explained: in vitro models are cheaper and have an increased throughput [73]. However, the user will always have to weigh up which test is selected for the purpose including the best significance for predicting human data. For pulmonary development, we suggest a resources- and benefit-driven approach. Resources are all efforts required for the performance of the selected in vitro assay. Benefit includes the prediction of human data and chances for regulatory acceptance, including the status of its validation. Putting this information in a two-dimensional x–y diagram with x = benefit and y = resources, we achieve four quadrants from which we can use the first three clockwise for developing orally inhaled drug products (Fig. 2). By passing this way to advanced testing, drug formulation candidates should decrease, and the targeted formulation will be optimized.

Benefits versus resources during an in vitro method development. Starting from the basic understanding to screening assays ending to advanced testing, the reduction of complexity increases to support the optimization of in vitro assays. In addition, the in vitro method, which gives no benefit, will be directly identified and no longer pursued
Table 2 summarizes in vitro models classified according to their complexity, use of resources and benefits. However, the predictability for in vivo outcomes of the here presented in vitro lung models, especially for predicting the kinetics of absorption and permeability, is still a subject of discussion [74].
To increase the predictivity, epithelial cells can be co-cultured or even triple-cultured with immune cells (THP-1, monocyte-derived macrophages (MDM)) and human pulmonary microvascular endothelial cells (HPMEC) to more accurately simulate the air-blood barrier [86,87,88,89,90,91]. Because the lung is constituted of many cellular and non-cellular (mucus, surfactant) barriers, whose spatial arrangement is essential for a functioning lung physiology, lung organoids were developed. Organoids are defined as 3D in vitro tissue structures simulating a complete in vivo organ [92, 93]. Most organoids are cultivated in a Matrigel matrix and are mostly generated from hPSC (human pluripotent stem cells) [94,95,96,97]. The main limitation of organoids is the low standardization of cell cultivation due to the complicated differentiation protocols with countless variations, which causes high costs [98]. Lung-on-a-chip models provide the cell cultivation in a microfluidic device. With a dynamic flow for media transport, a simulation of the function and exposure scenarios of the different lung areas is possible depending on the integrated cell types. However, this technology is still in its infancy [99,100,101,102]. A further development of complex in vitro lung systems is the 3D tissue cultures, which are commercially available, for example from MatTek and Epithelix. These systems include ciliated cells, goblet cells with a mucus layer and basal cells in the variant for the upper airways (MucilAir™, Epithelix) or the lower airways (SmallAir™, Epithelix), mucuciliary epithelium on fibroblasts (EpiAirway, MatTek), or the 3D constitution of alveolar epithelial cells, fibroblasts and alveolar endothelial cells (EpiAlveolar™, MatTek) [103,104,105,106]. The overall advantage of these ready-to-use systems is the high degree of standardization which is demonstrated by their presence in the ECVAM validation process (EpiAirway™) as mentioned above [72]. Further advantages of these systems are the individual user adaption through various cells (e.g. immune cells), the availability of diseased models and the combination with innovative exposure systems [107]. Primavessy et al. summarized the advantages of commercially available exposure systems which enhance the quality of in vitro systems due to a physiological lung simulation and substance deposition in comparison to static experimental setups [108]. One of the most renowned manufacturers of exposure systems for inhalation toxicology is VITROCELL® Systems. These VITROCELL® devices can expose airborne particles or chemicals (gases, mixtures, NPs) to many formats of cell cultures cultivated in liquid–liquid interface (LLI) or in air–liquid interface (ALI) [109]. In 2019, Kooter et al. investigated asthma patients’ increased sensitivity to copper oxide nanoparticle aerosols using the MucilAir™ (Epithelix) and a VITROCELL® exposure system. They observed a changed response of the model in a diseased state after NP exposure in comparison to the healthy cells by performing a transcriptomic analysis [110]. This experimental setup tries to simulate the particle concentration in the atmosphere more realistically than the calculation of particle concentration in dilution (LLI) [111].
Nevertheless, complex exposure experiments are still limited, due to technical restrictions as to simulating the highest dose of airborne particles and substances compared with surrounding contaminant concentration [112]. Future challenges for the standardization of physiologically relevant exposure systems with ALI cell cultures are the validation of in vitro to in vivo outcomes, the simulation of chronical exposure and the evaluation of the dosimetry (gas versus particles) to identify the exact NOAEL/LOAEL [112, 113].
These challenges were discussed during an international workshop with respiratory toxicology experts in 2018, which evaluated the necessary conditions and setup for an in vitro test system for respiratory safety assessment aiming the ECVAM validation. During the discussion, the main question arose as to which results can be validated to which endpoints regarding, for example, respiratory irritation, sensitization or inflammation. The problem being that no standardization of the established in vitro methods is possible, and, most critically, in vitro models are not compared to human clinical data and only correlated to animal data [68]. Based on these challenges, further research and additional data are still required for the improvement, validation and regulatory acceptance of respiratory in vitro models in order to establish them as alternatives to animal testing and to widen the implementation of the Three Rs principle in pharmaceutical research and industry.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current review article.
References
European Union, Directive 2010/63/EU of the European parliament and of the council of 22 September 2010 on the protection of animals used for scientific purposes, 2010 https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32010L0063. Accessed 26-Aug-2021.
Taylor K. Animal Experimentation: Working Towards a Paradigm Change. Brill, 2019.
European Commission, Ban on animal testing, 2021. https://ec.europa.eu/growth/sectors/cosmetics/animal-testing_en. Accessed: 17-Jan-2021.
European Union, Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products, 2009. https://eur-lex.europa.eu/eli/reg/2009/1223/oj. Accessed: 09-Jan-2021.
Van Norman GA. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic to Transl Sci. 2019;4(7):845–54. https://doi.org/10.1016/j.jacbts.2019.10.008.
Keen J. Animal Experimentation: Working Towards a Paradigm Change. 2019.
Meigs L, Smirnova L, Rovida C, Leist M, Hartung T. Animal testing and its alternatives - the most important omics is economics. ALTEX. 2018;35(3):275–305. https://doi.org/10.14573/altex.1807041.
European Commission, First report fom the commission to the council and the European parliament on the statistics on the number of animal experiments used for experimental and other scientific purposes. 1994.
European Commission, Report from the Commission to the European Parliament and the Council 2019: report on the statistics on the use of animals for scientific purposes in the Member States of the European Union in 2020:2015–2017.
Uhl EW, Warner NJ. Mouse Models as Predictors of Human Responses: Evolutionary Medicine. Curr Pathobiol Rep. 2015;3(3):219–23. https://doi.org/10.1007/s40139-015-0086-y.
Goh JY, Weaver RJ, Dixon L, Platt NJ, Roberts RA. Development and use of in vitro alternatives to animal testing by the pharmaceutical industry 1980–2013. Toxicol Res (Camb). 2015;4(5):1297–307. https://doi.org/10.1039/C5TX00123D.
Grand View research, Market Analysis Report, 2021. https://www.grandviewresearch.com/industry-analysis/in-vitro-toxicology-testing-market. Accessed: 26-Aug-2021.
Umweltbundesamt, REACH Was ist das? 2021. https://www.umweltbundesamt.de/themen/chemikalien/reach-chemikalien-reach. Accessed: 09-Jan-2021.
OECD (Organisation for Economic Cooperation and Development), OECD About, 2021. https://www.oecd.org/about/. Accessed: 11-Jan-2021.
OECD (Organisation for Economic Cooperation and Development), Test guidelines for chemicals. https://www.oecd.org/env/ehs/testing/oecdguidelinesforthetestingofchemicals.htm. Accessed: 05-Jan-2021.
OECD (Organisation for Economic Cooperation and Development), OECD Guidelines for the Testing of Chemicals, Section 4. https://www.oecd-ilibrary.org/environment/oecd-guidelines-for-the-testing-of-chemicals-section-4-health-effects_20745788. Accessed: 09-Jan-2021.
ECHA (European Chemicals Agency), Registration process. https://echa.europa.eu/de/support/dossier-submission-tools/reach-it/registration. Accessed: 18-Aug-2021.
Yusof I, Shah F, Hashimoto T, Segall MD, Greene N. Finding the rules for successful drug optimisation. Drug Discov Today. 2014;19(5):680–7. https://doi.org/10.1016/j.drudis.2014.01.005.
Lee D, Stein E, Gooneratne N. Overview of Drug Development. Acad Entrep Med Heal Sci. 2019;1(1):SBIR/STTR Grants: Introduction and Overview.
Owen K. Regulatory toxicology considerations for the development of inhaled pharmaceuticals. Drug Chem Toxicol. 2013;36(1):109–18. https://doi.org/10.3109/01480545.2011.648327.
Food and drug administration (FDA), Nonclinical Safety Evaluation of Reformulated Drug Products and Products Intended for Administration by an Alternate Route, 2008. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/nonclinical-safety-evaluation-reformulated-drug-products-and-products-intended-administration. Accessed 26-Aug-2021.
Food and Drug Administration (FDA), New Drug Development and Review Process, 2020. https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/new-drug-development-and-review-process. Accessed: 03-Jan-2021.
Chiodin D, Cox EM, Edmund AV, Kratz E, Lockwood SH. Regulatory Affairs 101: Introduction to Investigational New Drug Applications and Clinical Trial Applications. Clin Transl Sci. 2019;12(4):334–42. https://doi.org/10.1111/cts.12635.
Van Norman GA. Drugs and Devices: Comparison of European and U.S. Approval Processes. JACC Basic to Transl Sci. 2016;1(5):399–412. https://doi.org/10.1016/j.jacbts.2016.06.003.
Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586(7830):516–27. https://doi.org/10.1038/s41586-020-2798-3.
Srivastava G, Winslow A. Orphan Drugs : Understanding the FDA Approval Process, Acad Entrep Med Heal Sci, 2019;1 3.
Meekings KN, Williams CSM, Arrowsmith JE. Orphan drug development: An economically viable strategy for biopharma R&D. Drug Discov Today. 2012;17(13–14):660–4. https://doi.org/10.1016/j.drudis.2012.02.005.
V. Agarwall, Complete Guide on IND Enabling Toxicology Studies, 2020. https://www.nebiolab.com/complete-guide-on-ind-enabling-toxicology-studies. Accessed: 05-Jan-2021.
Food and Drug Administration (FDA), IND Forms and Instructions, 2017. https://www.fda.gov/drugs/investigational-new-drug-ind-application/ind-forms-and-instructions. Accessed: 05-Jan-2021.
Holbein ME. Understanding FDA Regulatory Requirements for Investigational New Drug Applications for Sponsor-Investigators. J Investig Med. 2009;57(6):688–94. https://doi.org/10.2310/JIM.0b013e3181afdb26.
Food and Drug Administration (FDA), CFR - Code of Federal Regulations Title 21 Protection of Human Subjects, 2015. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=50&showFR=1&subpartNode=21:1.0.1.1.20.2. Accessed: 05-Jan-2021
Andrade EL, et al. Non-clinical studies in the process of new drug development - Part II: Good laboratory practice, metabolism, pharmacokinetics, safety and dose translation to clinical studies. Brazilian J Med Biol Res. 2016;49(12): e5646. https://doi.org/10.1590/1414-431X20165646.
Food and Drug Administration (FDA), Guidance for Industry Process Validation: General Principles and Practices, 2011. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/process-validation-general-principles-and-practices. Accessed: 26-Aug-2021.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, Q7: Good manufacturing practice guide for active pharmaceutical ingredients, ICH Guidel., 2000.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH Guidelines, 2021. https://www.ich.org/page/ich-guidelines. Accessed: 06-Jan-2021.
EMEA, Non-clinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals: ICH M3 and M3(R2), 2013;3(July): 299–309.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Ich Harmonised Tripartite Guideline Pharmaceutical Development Q8(R2), Int Conf Harmon, 2009;3(June): 1–24.
van Dijk J, Gustavsson M, Dekker SC, van Wezel AP. Towards ‘one substance – one assessment’: An analysis of EU chemical registration and aquatic risk assessment frameworks. J Environ Manage. 2020;280(October 2020):111692. https://doi.org/10.1016/j.jenvman.2020.111692.
ECHA, The Use of Alternatives to Testing on Animals for the REACH Regulation. Third report under Article 117(3) of the REACH Regulation, 2017; 117(3).
Taylor K, Stengel W, Casalegno C, Andrew D. Experiences of the REACH testing proposals system to reduce animal testing. ALTEX. 2014;31(2):107–28. https://doi.org/10.14573/altex.1311151.
World Health Organization, The top 10 causes of death, 2020. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death. Accessed: 02-Jan-2021.
World Health Organization, WHO reveals leading causes of death and disability worldwide: 2000–2019, 2020. https://www.who.int/news/item/09-12-2020-who-reveals-leading-causes-of-death-and-disability-worldwide-2000-2019. Accessed: 03-Jan-2021.
Speizer FE, Horton S, Batt J, Slutsky AS, Chapter 35. Respiratory Diseases of Adults, Dis. Control Priorities Dev. Ctries. 2nd Ed. 2006;681–694.
Cookson WOCM, Cox MJ, Moffatt MF. New opportunities for managing acute and chronic lung infections. Nat Rev Microbiol. 2018;16(2):111–20. https://doi.org/10.1038/nrmicro.2017.122.
Janssens T, Ritz T. Perceived triggers of asthma: Key to symptom perception and management. Clin Exp Allergy. 2013;43(9):1000–8. https://doi.org/10.1111/cea.12138.
Linden D, et al. Respiratory viral infection: A potential ‘missing link’ in the pathogenesis of COPD. Eur Respir Rev. 2019;28(151):1–14. https://doi.org/10.1183/16000617.0063-2018.
McCracken GH. Etiology and treatment of pneumonia. Pediatr Infect Dis J. 2000;19(4):373–7. https://doi.org/10.1097/00006454-200004000-00032.
Villar J, Pérez-Méndez L, Kacmarek RM. Current definitions of acute lung injury and the acute respiratory distress syndrome do not reflect their true severity and outcome. Intensive Care Med. 1999;25(9):930–5. https://doi.org/10.1007/s001340050984.
Rezoagli E, Fumagalli R, Bellani G. Definition and epidemiology of acute respiratory distress syndrome. Ann Transl Med. 2017;5(14):1–12. https://doi.org/10.21037/atm.2017.06.62.
Johnson ER, Matthay M. Acute Lung Injury: Epidemiology, Pathogenesis, and Treatment. J Aerosol Med Pulm Drug Deliv. 2010;23(4):243–52. https://doi.org/10.1089/jamp.2009.0775.
Woodhead M, et al. Guidelines for the management of adult lower respiratory tract infections. Eur Respir J. 2005;26(6):1138–80. https://doi.org/10.1183/09031936.05.00055705.
Vestbo J, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–65. https://doi.org/10.1164/rccm.201204-0596PP.
Vestbo J et al. GOLD 2013 supplement. Am J Respir Crit Care Med. 2013;187(4): p. online supplement. https://doi.org/10.1164/rccm.201204-0596PP
Silva PL, Pelosi P, Rocco PRM. Personalized pharmacological therapy for ARDS: a light at the end of the tunnel. Expert Opin Investig Drugs. 2020;29(1):49–61. https://doi.org/10.1080/13543784.2020.1699531.
Qu JM, Zhu YG, Zhang J, Jiang HN, Xu JF, Novel interventional approaches for ALI/ARDS: Cell-based gene therapy, Mediators Inflamm, 2011;2011. https://doi.org/10.1155/2011/560194
Horie S, et al. Emerging pharmacological therapies for ARDS: COVID-19 and beyond. Intensive Care Med. 2020;46(12):2265–83. https://doi.org/10.1007/s00134-020-06141-z.
European Medical Agency, Orphan designation EU/3/20/2301, 2020. https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3202301. Accessed: 03-Jan-2021.
Bayer Pharmaceuticals, Develoment Pipeline, 2020. https://pharma.bayer.com/development-pipeline. Accessed: 03-Jan-2021.
Smaldone G, Berkland C, Gonda I, Mitchell J, Usmani OS, Clark A. Ask the experts: the benefits and challenges of pulmonary drug delivery. Ther Deliv. 2013;4(8):905–13.
Bains W, Failure Rates in Drug Discovery and Development-Will We Ever Get Any Better? Drug Discov World, 2004;9–18.
Corsini E, Papale A, Galbiati V, Roggen EL. Safety evaluation of cosmetic ingredients: In vitro opportunities for the identification of contact allergens. Cosmetics. 2014;1(1):61–74. https://doi.org/10.3390/cosmetics1010061.
Urbisch D, et al. Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol. 2015;71(2):337–51. https://doi.org/10.1016/j.yrtph.2014.12.008.
Urbisch D, et al. Peptide reactivity associated with skin sensitization: The QSAR Toolbox and TIMES compared to the DPRA. Toxicol Vitr. 2016;34:194–203. https://doi.org/10.1016/j.tiv.2016.04.005.
European Chemicals Agency, How to use new or revised in vitro test methods to address skin sensitisation, 2018;no. February, pp. 1–11.
OECD (Organisation for Economic Cooperation and Development), Section 4: Health Effects, OECD Guidel. Test. Chem., 2018;no. June, 16.
EURL. ECVAM, Validated test methods. https://ec.europa.eu/jrc/en/eurl/ecvam/alternative-methods-toxicity-testing/validated-test-methods. Accessed: 26-Aug-2021.
OECD, Guidance Document on Good In Vitro Method Practices (GIVIMP), 2018;no. 286.
Lacroix G, et al. Air-Liquid Interface in Vitro Models for Respiratory Toxicology Research: Consensus Workshop and Recommendations. Appl Vitr Toxicol. 2018;4(2):91–106. https://doi.org/10.1089/aivt.2017.0034.
Bakand S, Winder C, Khalil C, Hayes A. Toxicity assessment of industrial chemicals and airborne contaminants: Transition from in vivo to in vitro test methods: A review. Inhal Toxicol. 2005;17(13):775–87. https://doi.org/10.1080/08958370500225240.
US Department of human health, About ICCVAM, 2021. Available: https://ntp.niehs.nih.gov/whatwestudy/niceatm/iccvam/index.html. Accessed: 17-Jan-2021.
European Commission, EU Reference Laboratory for alternatives to animal testing. https://ec.europa.eu/jrc/en/eurl/ecvam. Accessed: 17-Jan-2021.
E. ECVAM, Jrc Science for Policy Report- EURL ECVAM Status Report on the Development, Validation and Regulatory Acceptance of Alternative Methods and Approaches. 2019.
Hittinger M, Juntke J, Kletting S, Schneider-Daum N, de Souza Carvalho C, Lehr CM. Preclinical safety and efficacy models for pulmonary drug delivery of antimicrobials with focus on in vitro models. Adv Drug Deliv Rev. 2015;85:44–56.
Sakagami M. In vivo, in vitro and ex vivo models to assess pulmonary absorption and disposition of inhaled therapeutics for systemic delivery. Adv Drug Deliv Rev. 2006;58(9–10):1030–60. https://doi.org/10.1016/j.addr.2006.07.012.
Sou T, Bergström CAS. Contemporary Formulation Development for Inhaled Pharmaceuticals. J Pharm Sci. 2021;110(1):66–86. https://doi.org/10.1016/j.xphs.2020.09.006.
United States Pharmacopeial Convention. USP <601> Physical tests and determinations: Aerosols. Nasal sprays, Metered-dose inhalers, and dry powder inhalers. Pharmacopeial Forum. 2009;35(3):615–28.
Metz J, et al. Safety assessment of excipients (SAFE) for orally inhaled drug products. ALTEX. 2020;1(2):275–86. https://doi.org/10.14573/altex.1910231.
Scherließ R. The MTT assay as tool to evaluate and compare excipient toxicity in vitro on respiratory epithelial cells. Int J Pharm. 2011;411(1–2):98–105. https://doi.org/10.1016/j.ijpharm.2011.03.053.
Sauer UG, et al. In vivo-in vitro comparison of acute respiratory tract toxicity using human 3D airway epithelial models and human A549 and murine 3T3 monolayer cell systems. Toxicol Vitr. 2013;27(1):174–90. https://doi.org/10.1016/j.tiv.2012.10.007.
Meindl C, et al. Permeation of therapeutic drugs in different formulations across the airway epithelium in vitro. PLoS ONE. 2015;10(8):1–19. https://doi.org/10.1371/journal.pone.0135690.g004.
Hoppstädter J, et al. Differential cell reaction upon Toll-like receptor 4 and 9 activation in human alveolar and lung interstitial macrophages. Respir Res. 2010;11:1–15. https://doi.org/10.1186/1465-9921-11-124.
Juntke J, et al. Testing of aerosolized ciprofloxacin nanocarriers on cystic fibrosis airway cells infected with P. aeruginosa biofilms. Drug Deliv Transl Res. 2021;11(4):1752–65. https://doi.org/10.1007/s13346-021-01002-8.
Pizzorno A et al., Characterization and Treatment of SARS-CoV-2 in Nasal and Bronchial Human Airway Epithelia, Cell Reports Med., 2020;1(4). https://doi.org/10.1016/j.xcrm.2020.100059
Giralt A, et al. Comparison of the biological impact of aerosol of e-vapor device with MESH® technology and cigarette smoke on human bronchial and alveolar cultures. Toxicol Lett. 2021;337:98–110. https://doi.org/10.1016/j.toxlet.2020.11.006.
Marescotti D, et al. How complex should an in vitro model be? Evaluation of a complex 3D alveolar model with transcriptomic data and computational biological network models. ALTEX. 2019;36(3):388–402. https://doi.org/10.14573/altex.1811221.
Mangum JB, Everitt JI, Bonner JC, Moore LR, Brody AR. Co-culture of primary pulmonary cells to model alveolar injury and translocation of proteins. Vitr Cell Dev Biol. 1990;26(12):1135–43. https://doi.org/10.1007/BF02623690.
Hittinger M, Janke J, Huwer H, Scherließ R, Schneider-Daum N, Lehr CM. Autologous co-culture of primary human alveolar macrophages and epithelial cells for investigating aerosol medicines. Part I: Model Characterisation. ATLA Altern to Lab Anim. 2016;44(4):337–47. https://doi.org/10.1177/026119291604400404.
Hittinger M, Mell NA, Huwer H, Loretz B, Schneider-Daum N, Lehr CM. Autologous co-culture of primary human alveolar macrophages and epithelial cells for investigating aerosol medicines. Part II: Evaluation of IL-10-loaded microparticles for the treatment of lung inflammation. ATLA Altern to Lab Anim. 2016;44(4):349–60. https://doi.org/10.1177/026119291604400405.
Kletting S et al., Co-culture of human alveolar epithelial, 2017;1–11. https://doi.org/10.14573/altex.1607191
Lehmann AD, Daum N, Bur M, Lehr CM, Gehr P, Rothen-Rutishauser BM. An in vitro triple cell co-culture model with primary cells mimicking the human alveolar epithelial barrier. Eur J Pharm Biopharm. 2011;77(3):398–406. https://doi.org/10.1016/j.ejpb.2010.10.014.
Costa A, de Souza Carvalho-Wodarz C, Seabra V, Sarmento B, Lehr CM. Triple co-culture of human alveolar epithelium, endothelium and macrophages for studying the interaction of nanocarriers with the air-blood barrier. Acta Biomater. 2019;91:235–47. https://doi.org/10.1016/j.actbio.2019.04.037.
De Souza N. Organoids. Nat Methods. 2018;15(1):23. https://doi.org/10.1038/nmeth.4576.
Barkauskas CE, Chung MI, Fioret B, Gao X, Katsura H, Hogan BLM. Lung organoids: Current uses and future promise. Dev. 2017;144(6):986–97. https://doi.org/10.1242/dev.140103.
Miller AJ, et al. Generation of lung organoids from human pluripotent stem cells in vitro. Nat Protoc. 2019;14(2):518–40. https://doi.org/10.1038/s41596-018-0104-8.
Barkauskas CE, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025–36.
Youk J, et al. Three-Dimensional Human Alveolar Stem Cell Culture Models Reveal Infection Response to SARS-CoV-2. Cell Stem Cell. 2020;27(6):905-919.e10.
Baptista D, et al. 3D alveolar in vitro model based on epithelialized biomimetically curved culture membranes. Biomaterials. 2020;266(October):2021. https://doi.org/10.1016/j.biomaterials.2020.120436.
Kim J, Koo BK, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. 2020;21(10):571–84. https://doi.org/10.1038/s41580-020-0259-3.
Shrestha J, et al. Lung-on-a-chip: the future of respiratory disease models and pharmacological studies. Crit Rev Biotechnol. 2020;40(2):213–30.
Shrestha J, et al. A rapidly prototyped lung-on-a-chip model using 3D-printed molds. Organs-on-a-Chip. 2019;1:100001. https://doi.org/10.1016/j.ooc.2020.100001.
Stucki AO, et al. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip. 2015;15(5):1302–10. https://doi.org/10.1039/C4LC01252F.
Benam KH, et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods. 2016;13(2):151–7. https://doi.org/10.1038/nmeth.3697.
Cervena T, Vrbova K, Rossnerova A, Topinka J, Rossner P. Short-term and Long-term Exposure of the MucilAirTM Model to Polycyclic Aromatic Hydrocarbons. Altern Lab Anim. 2019;47(1):9–18.
Huang S, Boda B, Vernaz J, Ferreira E, Wiszniewski L, Constant S. Establishment and characterization of an in vitro human small airway model (SmallAirTM). Eur J Pharm Biopharm. 2017;118:68–72. https://doi.org/10.1016/j.ejpb.2016.12.006.
Mistry A, Bowen LE, Dzierlenga MW, Hartman JK, Slattery SD. Development of an in vitro approach to point-of-contact inhalation toxicity testing of volatile compounds, using organotypic culture and air-liquid interface exposure. Toxicol Vitr. 2020;69:104968. https://doi.org/10.1016/j.tiv.2020.104968.
Barosova H, et al. Use of EpiAlveolar Lung Model to Predict Fibrotic Potential of Multiwalled Carbon Nanotubes. ACS Nano. 2020;14(4):3941–56. https://doi.org/10.1021/acsnano.9b06860.
Cao X, et al. Invited review: human air-liquid-interface organotypic airway tissue models derived from primary tracheobronchial epithelial cells—overview and perspectives. Vitr Cell Dev Biol - Anim. 2020. https://doi.org/10.1007/s11626-020-00517-7.
Primavessy D, Metz J, Schnur S, Schneider M, Lehr C-M, Hittinger M, Pulmonary in vitro instruments for the replacement of animal experiments. Eur Pharm Biopharm, 2021.Accepted 24-Aug-2021.
Vitrocell Systems, Exposure systems for inhalation toxicology. https://www.vitrocell.com/inhalation-toxicology/exposure-systems. Accessed: 23-Jan-2021.
Kooter I, et al. Molecular Signature of Asthma-Enhanced Sensitivity to CuO Nanoparticle Aerosols from 3D Cell Model. ACS Nano. 2019. https://doi.org/10.1021/acsnano.9b01823.
L. Müller et al., Realistic Exposure Methods for Investigating the Interaction of Nanoparticles with the Lung at the Air-Liquid Interface In Vitro. Insciences J. 2011;2014: 30–64.
Upadhyay S, Palmberg L. Air-liquid interface: Relevant in vitro models for investigating air pollutant-induced pulmonary toxicity. Toxicol Sci. 2018;164(1):21–30. https://doi.org/10.1093/toxsci/kfy053.
Schmid O, Cassee FR. On the pivotal role of dose for particle toxicology and risk assessment: exposure is a poor surrogate for delivered dose. Part Fibre Toxicol. 2017;14(1):52. https://doi.org/10.1186/s12989-017-0233-1.
OECD (Organisation for Economic Cooperation and Development), “Test guidelines 403 acute inhalation toxicity,” 2009;11.
OECD (Organisation for Economic Cooperation and Development), Test guideline 433 acute inhalation toxicity: fixed concentration procedure, 2018;39.
OECD (Organisation for Economic Cooperation and Development), Test guideline 436 acute inhalation toxicity - acute toxic class method, 2009.
OECD (Organisation for Economic Cooperation and Development), Test guideline 412 28-day (subacute) inhalation toxicity study, 39, no. June, 2018.
OECD (Organisation for Economic Cooperation and Development), Test guideline 413 90-Day subchronic inhalation toxicity study, 2018.
Cheek JM, Evans MJ, Crandall ED. Type I cell-like morphology in tight alveolar epithelial monolayers. Exp Cell Res. 1989;184(2):375–87. https://doi.org/10.1016/0014-4827(89)90337-6.
Foster KA, Oster CG, Mayer MM, Avery ML, Audus KL. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp Cell Res. 1998;243(2):359–66. https://doi.org/10.1006/excr.1998.4172.
Kobayashi S, Shuji K, Juni K. Permeability of Peptides and Proteins in human cultured alveolar A549 cell monolayer. Pharm Res. 1995;12(8):1115–9. https://doi.org/10.1023/A:1016295406473.
C-M. Lehr, Elbert, Katharina J, Schäfer Ulrich F., Schäfers, Hans-Joachim, Kim, Kwang-Jin, Monolayers of human alveolar epithelial cells in primary culture for pulmonary absorption and transport studies, Pharm Research, 1999;16(5). https://doi.org/10.1023/A:1018887501927.
Schneider-Daum et al. Isolation, Cultivation, and Application of Human Alveolar Epithelial Cells. Hum Cell Cult Protoc. 2012;806. https://doi.org/10.1007/978-1-61779-367-7_3.
Kuehn A, et al. Human alveolar epithelial cells expressing tight junctions to model the air-blood barrier. ALTEX. 2016;33(3):251–60. https://doi.org/10.14573/altex.1511131.
Inscrennex, Alveolar Epithelial Cells, 2021. https://www.inscreenex.de/products/human-immortalized-cell-lines/alveolar-epithelial-cells-hu.html. Accessed: 22-Jan-2021.
van Riet S, et al. In vitro modelling of alveolar repair at the air-liquid interface using alveolar epithelial cells derived from human induced pluripotent stem cells. Sci Rep. 2020;10(1):1–12. https://doi.org/10.1038/s41598-020-62226-1.
Acknowledgements
We thank Julia Petry for her help in proofreading the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Julia Metz was financially supported by the BMBF project AeroSafe (031L0128C). Dr. Marius Hittinger was financially supported by the BMWi INNO-KOM project Kombipulmonal (49MF200045).
Author information
Authors and Affiliations
Contributions
Conceptualization: Julia Metz, Marius Hittinger, Claus-Michael Lehr. Writing original draft preparation: Julia Metz. Writing—review and editing: Marius Hittinger, Claus-Michael Lehr. Supervision: Marius Hittinger, Claus-Michael Lehr.
Corresponding author
Ethics declarations
Ethics approval
No approval of research ethics committees was required to accomplish the goals of this study because no experimental work was conducted with an unregulated invertebrate species.
Conflict of interest
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Metz, J.K., Hittinger, M. & Lehr, CM. In vitro tools for orally inhaled drug products—state of the art for their application in pharmaceutical research and industry and regulatory challenges. In vitro models 1, 29–40 (2022). https://doi.org/10.1007/s44164-021-00003-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s44164-021-00003-8