
Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1


1
Genomic Instability Group, Spanish National Cancer Research Centre (CNIO), 28029 Madrid, Spain
2
Science for Life Laboratory, Division of Genome Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institute, S-171 21 Stockholm, Sweden
3
Centre for Molecular Biology Severo Ochoa (CBMSO, CSIC-UAM), Chromatin, Cancer and the Ubiquitin System Lab, Department of Genome Dynamics and Function, 28049 Madrid, Spain
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally.
Int. J. Mol. Sci. 2021, 22(16), 8796; https://doi.org/10.3390/ijms22168796
Submission received: 20 July 2021
/
Revised: 11 August 2021
/
Accepted: 12 August 2021
/
Published: 16 August 2021
(This article belongs to the Special Issue The Role of Post-translational Modifications in Chromatin and Cancer)
Abstract
:Post-translational modification of the DNA replication machinery by ubiquitin and SUMO plays key roles in the faithful duplication of the genetic information. Among other functions, ubiquitination and SUMOylation serve as signals for the extraction of factors from chromatin by the AAA ATPase VCP. In addition to the regulation of DNA replication initiation and elongation, we now know that ubiquitination mediates the disassembly of the replisome after DNA replication termination, a process that is essential to preserve genomic stability. Here, we review the recent evidence showing how active DNA replication restricts replisome ubiquitination to prevent the premature disassembly of the DNA replication machinery. Ubiquitination also mediates the removal of the replisome to allow DNA repair. Further, we discuss the interplay between ubiquitin-mediated replisome disassembly and the activation of CDK1 that is required to set up the transition from the S phase to mitosis. We propose the existence of a ubiquitin–CDK1 relay, where the disassembly of terminated replisomes increases CDK1 activity that, in turn, favors the ubiquitination and disassembly of more replisomes. This model has important implications for the mechanism of action of cancer therapies that induce the untimely activation of CDK1, thereby triggering premature replisome disassembly and DNA damage.
1. The Ubiquitin and SUMO System
Post-translational modifications (PTMs) play key roles in the cell as essential regulators of the spatiotemporal control of protein function. Among the PTMs, ubiquitin (Ub) is a small protein that is conjugated to lysine residues on target proteins and mark them for degradation by the proteasome, a process that was discovered 40 years ago [1]. Ubiquitination is a three-step process, where ubiquitin is first conjugated to the E1 activating enzyme, then transferred to an E2 conjugating enzyme and finally attached to a specific substrate through the cooperation of ubiquitin E3 ligases and their partner, E2. The system achieves great specificity thanks to the existence of more than 600 E3 ligases in the human genome. SUMO (small ubiquitin-like modifier) is a 100 amino acid ubiquitin-like protein that was discovered in the 1990s and displays high homology with ubiquitin [2]. The SUMOylation pathway also involves a single E1 enzyme that transfers SUMO to UBC9, the only E2 enzyme [3,4]. UBC9 transfers SUMO to its targets by itself or in association with a limited number of specific E3 SUMO ligases [5].
The ubiquitin system can generate a slew of different configurations from a single ubiquitin conjugated to one or several lysine residues (monoubiquitination and multi-monoubiquitination) or ubiquitin chains, either linear or branched. In mammals, SUMO2/3 also forms chains and can also participate in mixed ubiquitin/SUMO chains. This “ubiquitin code” establishes a set of specific modifications with different functional outcomes by modifying the stability, localization, activity or interactome of the target proteins. One of the main transducers of ubiquitin and SUMO is the AAA ATPase VCP (valosin containing protein), also known as p97 segregase, a central factor in the regulation of protein homeostasis [6,7,8,9] that mobilizes the chromatin-bound substrates conjugated to ubiquitin [10,11,12] or ubiquitin-like molecules [13,14,15,16]. To this end, VCP uses a repertoire of adaptors/cofactors which recognize poly-ubiquitinated substrates through their ubiquitin-binding domains [17,18]. These cofactors associate with VCP in a hierarchical system, where the primary cofactors determine the binding of additional adaptors to determine specific VCP–adaptor configurations that recognize the different substrates [19,20].
Both ubiquitination and SUMOylation can be reverted by the action of specific proteases called deubiquitinases (DUBs) [21] and SUMO-specific proteases [22]. Finally, there is crosstalk between ubiquitination and SUMOylation with a specialized set of E3 ubiquitin ligases, known as SUMO-targeted ubiquitin ligases (STUbLs), which recognize and ubiquitinate SUMOylated proteins [23]. Conversely, some DUBs have been shown to be active on SUMO and SUMOylated proteins, including USP7 [24] and USP11 [25]. Ub and SUMO constitute a complex post-translational modification system that affects many essential cellular functions, including the regulation of DNA metabolism and genome stability [26].
2. SUMO and Ubiquitin in DNA Replication
DNA replication mediates the accurate copy of the entire genetic material of a cell. Thus, it needs to be tightly regulated to preserve genome integrity and prevent the accumulation of DNA damage and replicating errors that lead to cancer development and the onset of aging. DNA replication can be divided into three phases: initiation, the S phase or elongation and DNA replication termination. The ubiquitin/SUMO pathways are essential regulators of DNA replication in each of these phases (Table 1).
DNA replication initiation is a two-step process involving origin licensing in G1 and the subsequent formation of the pre-replication complex (pre-RC) that is required to transition into the S phase. Origin licensing takes place in all potential replication origins by the sequential loading of two protein complexes. First, the origin recognition complex (ORC1-6) binds origins of replication. Next, the mini-chromosome maintenance (MCM) complex is recruited with the help of the ATPase CDC6 and CDT1 (CDC10-dependent transcript 1) to form the pre-RC [76]. However, only a subset of these origins is activated or fired in each S phase [77]. A recent report has identified OBI1 as an E3 ubiquitin ligase that multi-monoubiquitinates ORC3 and ORC5 to promote the firing of a subset of origins of replication without affecting the assembly of the pre-RC (Figure 1) [27]. To avoid re-replication of the DNA in the S phase, the origins need to be fired only once per cell cycle, and the ubiquitin system also prevents the re-licensing of replication origins after they have been fired. Both ORC1 and CDT1 are poly-ubiquitinated after the initiation of DNA replication by the SCFSkp2 ubiquitin–ligase complex (Figure 1). Consequently, they are extracted from chromatin by VCP and undergo proteasome-mediated degradation [28,29].
Once the pre-RC is assembled, origin activation marks the onset of the S phase by triggering the unwinding of the DNA double helix to allow loading of the replication machinery. Origin activation requires the phosphorylation of several subunits of the MCM complex by the Ser/Thr protein kinases DBF4-dependent kinase (DDK) and by interphase cyclin dependent kinases (CDKs) [78,79]. This phosphorylation promotes the assembly of the replicative CMG helicase through recruitment of the GINS complex and CDC45 [80]. The phosphorylation of the MCM complex is controlled by ubiquitin/SUMO in two steps (Figure 1). First, SUMOylation of MCM proteins limits their phosphorylation in G1, preventing premature origin activation [66]. In the transition to the S phase SUMOylation levels decline as the phosphorylation of the MCM complex rises. Second, the ubiquitin/SUMO pathways also switch off the origin activation signal by timing the degradation of the DDK. Chromatin-bound DDK is SUMOylated, leading to its ubiquitination by STUbLs to induce its degradation by the proteasome. The SUMO protease Ulp2 protects DDKs from degradation, allowing the early steps of DNA replication to occur [63].
After the origins have been activated, the double MCM2-7 hexamer divides and establishes two replication forks through the recruitment of additional replication factors including RPA, RFC, PCNA and the replicative DNA polymerases [77]. Proteomic analyses of chromatin under replication have revealed that DNA replication forks are embedded in a SUMO-rich environment [81,82,83]. Although the exact functions of replisome SUMOylation remain to be elucidated, a recent report showed that the SUMOylation of the catalytic subunit of DNA polymerase ε is important for DNA replication in yeast [64,65]. In addition to the SUMOylation of specific factors, we have proposed that the collective SUMOylation of the replication machinery supports DNA replication by creating an environment that facilitates interactions among replication factors [84], analogous to the SUMO-based group modification model proposed by Stefan Jentsch for DNA repair [85]. Within this model, we previously showed that USP7 is a SUMO-dependent deubiquitinase that maintains low levels of ubiquitination in the replisome, and whose action is necessary to sustain DNA replication [24]. This is particularly relevant in light of the recent advances showing how ubiquitin and SUMO are essential for disassembly of the replication machinery after DNA replication termination, as we develop in the next section.
3. DNA Replication Termination
The study of DNA replication termination is not technically easy and, as a consequence, its molecular mechanisms have remained relatively unexplored compared with the initiation and elongation phases [86,87,88]. Recent studies have shed light on the sequence of events that sets up the end of DNA replication when the two forks converge [89,90,91]. During fork convergence, the action of topoisomerases to release topological stress is impeded by the lack of space ahead of the forks [92]. Instead, this stress is relieved by the clockwise rotation of the two forks, which generates intertwining between the two replicated sister chromatids. This intertwining is finally resolved by Type II topoisomerases or by the action of Pif1 and Rrm3 helicases [93,94,95,96,97,98,99,100,101]. Thanks to these enzymes, the replisome encounter does not induce fork stalling, and the CMG helicases keep moving at the same speed to rapidly pass each other, moving from the leading to the lagging strand of the converging fork [102]. Next, the single-stranded gap between the 3ʹ end of the leading strand and the downstream Okazaki fragment of the opposing fork is filled in and the CMGs travel on dsDNA no longer supporting DNA synthesis but allowing Okazaki fragment processing by Polδ and FEN1 [102]. At this stage, the replication machinery is disassembled from dsDNA in a process that involves the ubiquitination of the CMG helicase and the action of the AAA ATPase VCP [102]. Finally, the sister chromatids need to be decatenated by Type II topoisomerases before chromosome segregation [103,104]. Evidence accumulated in recent years has shown that the disassembly of the replication machinery is one of the key steps in DNA replication termination and that the ubiquitin pathway lies at the heart of this process.
4. Replisome Disassembly at the End of DNA Replication
As CMGs cannot be reloaded during the elongation phase, removal of the replisome irreversibly blocks fork progression [105]. Thus, the eviction of the replication machinery is tightly regulated and restricted to replication forks that have reached the downstream Okazaki fragment to ensure full replication of the DNA [102]. The molecular events that control replisome disassembly are only beginning to be understood. One of the events associated with DNA replication termination is the poly-ubiquitination of the MCM7 subunit of the CMG helicase at lysine-48 (K48) that is recognized by VCP through the ubiquitin fusion degradation protein 1 homolog (UFD1L) and nuclear protein localization protein 4 homolog (NPLOC4) heterodimer [70,106,107]. It has been proposed that MCM7 ubiquitination helps in the extraction of many components of the replication machinery from chromatin (Figure 2A) [68,69,71,108]. Notably, while much of the attention has been placed on MCM7 ubiquitination on replisome disassembly during DNA replication, it seems unlikely that this is the only critical ubiquitination event that is involved in DNA replication termination. Many additional replisome components might be similarly ubiquitinated and extracted from chromatin following the same or equivalent pathways, and MCM7 ubiquitination may serve as a model to understand the mechanisms governing ubiquitin-mediated replisome eviction.
There are two basic mechanisms that control ubiquitination-driven replisome disassembly to avoid untimely eviction of the replication machinery: the first one is the spatio-temporal restriction of the action of E3 ubiquitin-ligases; the second one is the ubiquitin threshold for the action of VCP. The E3 ligase in charge of ubiquitinating MCM7 differs between yeast and higher eukaryotes. While yeast SCF (Skp, Cullin, F-box-containing complex) associates with Dia2 to induce MCM7 ubiquitination [68], in higher eukaryotes, the Cullin RING ligase 2 (CUL2) protein binds to LRR1 to modify MCM7 [69,70,71]. How is the activity of these E3 ligases restricted during DNA replication elongation? Recent work has shown that the presence of the lagging strand in active DNA replication forks inhibits the action of the E3 ligases or blocks their access to the replisome. Once the final parental duplex has unwound, the CMGs helicases move on to dsDNA and the interaction with the lagging strand is lost, allowing CMG ubiquitination [109,110]. In yeast, Dia2 interacts with Ctf4 and Mrc1 and it has been proposed that SCFDia2 travels together with the replication machinery [111,112,113,114]. The interaction of the lagging strand template with the CMG helicase would mask the substrates of SCFDia2, although a direct action preventing its association with the replisome has not been completely ruled out [110]. In the case of CUL2LRR1, the lagging strand directly prevents the interaction of its LRR domain with the CMG helicase, and thus it is specifically targeted to terminated CMGs [70,71]. The active DNA replication model could explain how a single helicase is disassembled at the end of a telomere or at a single-stranded nick, but it does not justify how CMG ubiquitination is prevented during DNA replication initiation before the replication forks have formed [115].
A second layer of control is established through the length of the ubiquitin chains on the CMG, since the action of VCP depends on a “ubiquitin threshold”, whereby VCP/Ufd1/Npl4 requires four to five ubiquitin molecules to act on its substrates [116]. In yeast, SCFDia2 only generates short ubiquitin chains on MCM7 during DNA replication, most likely due to the inhibition exerted by the interaction of the lagging strand with the CMG [110]. A recent report showed that TIMELESS-TIPIN stimulates the activity of CUL2LRR1 in C. elegans to achieve efficient ubiquitination of the CMG [117]. The ubiquitination of the CMG could also be limited by specific DUBs, and we have previously identified USP7 as a SUMO-specific deubiquitinase that maintains a SUMO-rich/ubiquitin-poor environment at the DNA replication forks [24,81]. In this sense, SUMO could act as a signal to drive the ubiquitination of replication factors upon DNA replication termination or compete for ubiquitination to prevent the untimely modification of these proteins [84]. In yeast, the MCM2-7 complex has been shown to be SUMOylated in G1, and MCM7 SUMOylation persists during the S phase [66]. Interestingly, proteomic analyses revealed that USP7 inhibition induces the ubiquitination of many replication factors, including MCM7, suggesting it may play a role in preventing the premature disassembly of the replisome [24]. On the other hand, USP7 was previously found to interact and cooperate with the MCM-binding protein (MCM-BP) in unloading MCM7 [118]. Thus, USP7 may have opposing actions during the disassembly of the replication machinery, and how these actions are coordinated has not been clarified yet.
The relevance of ubiquitin-mediated replisome disassembly was further substantiated by in vitro experiments in yeast. The minimal requirements for replisome disassembly involve the ubiquitination of the replication machinery, including MCM7, by SCFDia2 and its extraction by Cdc48 in combination with Ufd1/Npl4 [114]. Whether other components of the replication machinery are ubiquitinated and contribute to the recruitment of VCP and the eviction of specific proteins has not been explored yet. In addition, the interplay between MCM7 ubiquitination and SUMOylation remains to be studied. Once MCM7 has been ubiquitinated, little is known about its fate. Although VCP often targets proteins for their proteasomal degradation [7,119], the inhibition of the proteasome has no effect on MCM7 accumulation [120]. These data suggest that the CMG complex is recycled alongside other components of the replication machinery. It would be interesting to determine the fate of the replication factors upon replisome disassembly.
5. Replisome Disassembly after DNA Damage and outside the S Phase
Reinforcing the central role of ubiquitination for replisome disassembly, there are two situations outside the canonical pathway after DNA replication termination where MCM7 is also modified and contributes to the removal of the replication machinery from chromatin. First, if the canonical replisome extraction pathway fails, the replication machinery needs to be mobilized before mitosis. In contrast to yeast, work in C. elegans and mouse embryonic stem cells has revealed the existence of a back-up mechanism for replisome unloading in mitosis by an alternative pathway independent of CUL2LRR1 [69,70,73,74]. Mitotic replisome disassembly involves the TRAIP-dependent ubiquitination of MCM7 at K6 and K63 (Figure 2A) [69,73,74]. After its ubiquitination, MCM7 is extracted by VCP/Ufd1/Npl4 in cooperation with an additional cofactor, UBX domain-containing 3 (UBXN3; the worm ortholog of human FAS-associated factor 1 (FAF1)). The process requires the action of the SUMO protease ULP4 (the worm ortholog of SENP6/7), although its exact functions remain unknown [70] and SUMOylation does not affect this pathway in Xenopus egg extracts [74]. Similar to SCFDia2 in yeast, the E3 ligase TRAIP is constitutively associated with the replisome, where it plays a role during the repair of DNA–protein crosslinks [70,71,113,121]. The mechanisms controlling TRAIP activation in mitosis are still unknown. Abrogating both interphase and mitotic disassembly pathways by combined knockdown of LRR1 and UBXN3 stabilizes the presence of CMG on chromatin through the metaphase and leads to synthetical lethality [70].
Second, there are several types of DNA damage that require replisome disassembly to allow for repair during the S phase. Common fragile sites (CFSs) and other difficult-to-replicate regions are copied late in the S phase and often induce fork stalling, leading to replication stress and subsequently to an increase in anaphase ultrafine bridges, copy number variations or chromosomal rearrangements [122,123,124,125]. A recent work has linked the appearance of genomic alterations at CFS to premature disassembly of the replisome at these sites. This disassembly is associated with the ubiquitination of MCM7 by TRAIP (Figure 2B). After the eviction of the replication machinery, these structures are repaired by microhomology-mediated end joining (MMEJ), leading to the appearance of chromosomal alterations [73] similar to the ones observed in CFSs. Interestingly, the action of TRAIP at stalled forks is stimulated by CDK1, linking the activation of the mitotic program to the disassembly of the replisome.
Another instance that induces replisome extraction from chromatin is the presence of single-strand breaks (SSB) (Figure 2C). These breaks are often generated by problems during the action of topoisomerase I, and they are also produced as intermediates in base excision repair (BER) [126]. The repair mechanisms induced upon SSB-induced fork collapse are strand-specific but always involve the removal of CMG from chromatin [72]. When the break is present in the leading strand, the advance of the CMG helicase generates a single-ended double-strand break (seDSB), where the CMG passively slides off (Figure 2C). In contrast, when the break is present in the lagging strand, the fork generates a ssDSB with a single strand overhang while the CMG moves on the dsDNA. Following the same pathway that acts after DNA replication termination, the replication machinery is then ubiquitinated and evicted by VCP (Figure 2C) [69,70,71,109,110]. Last, DNA interstrand crosslinks (ICLs) covalently link both strands of the DNA molecule and block DNA replication and transcription [127]. There are two pathways for ICL repair mediated by the Fanconi anemia (FA) family of proteins and the NEIL3 glycosylase [128,129,130]. Both pathways involve the convergence of the two DNA replication forks and their stalling next to the lesion [131,132], followed by the ubiquitination of MCM7 at K48 by the E3 ligase TRAIP (Figure 2D) [75]. The length of the ubiquitin chains on MCM7 determines the repair pathway that is activated. Longer chains induce CMG unloading by the VCP segregase in both converging forks [75,120,133]. Despite the similarity to DNA replication termination, there is an additional contribution of BRCA1 in promoting CMG ubiquitination and unloading in response to ICL [120]. The removal of the replication machinery is necessary to allow the action of the FA machinery to repair the damage. On the other hand, short ubiquitin chains deposited on MCM7 at K48 by TRAIP activate the NEIL3 glycosylase pathway [75]. NEIL3 directly binds ubiquitinated MCM7 and mediates the unhooking of the ICL that does not require the disassembly of the replication machinery [134,135].
Thus, cells have evolved alternative pathways for replisome disassembly that work during mitosis or in the presence of DNA damage. All these pathways share the ubiquitination of the replication machinery as a central event, but they are mediated by specific mechanisms that involve, in many cases, the E3 ubiquitin ligase TRAIP. How TRAIP is specifically activated upon damage or during mitosis independently of the convergence of DNA replication forks and how its action is blocked during DNA replication termination [68,69,70,71] is still not understood.
6. The Ubiquitin Connection between DNA Replication and CDK1 Activation
The traditional view of the cell cycle considered DNA replication completion and the activation of the mitotic program as independent events separated by the G2 phase. However, there is increasing evidence to support a direct functional link between the end of the S phase and the onset of mitosis. First, the mitotic machinery is activated right after the PCNA foci disappear [136] and the active DNA replication forks suppress mitotic activity [137]. Based on these observations, the Lindqvist lab proposed a model for progression from the S phase to mitosis, where active DNA replication forks work as molecular breaks that prevent premature mitotic entry [138]. This model also implies the existence of a signal during DNA replication termination, when the active DNA replication forks disappear, that deactivates these breaks to start the mitotic program. Interestingly, CMG ubiquitination and replisome disassembly are also suppressed by active DNA replication forks, suggesting that this modification might link DNA replication termination and the progression into mitosis [109,110]. Conversely, the activation of CDK1 promotes the ubiquitination of the CMG and disassembly of the replisome [73]. Together, these observations support the existence of a ubiquitin–CDK1 relay, where the disassembly of terminated replisomes fosters CDK1 activation (Figure 3A). In turn, CDK1 activity reinforces disassembly of the replication machinery from terminated replisomes. Thus, replisome disassembly would not be favored in the early S phase when CDK1 activity is low. The increased CDK1 activity in the late S phase could drive the premature removal of the replication machinery from stalled forks at CFS and difficult-to-replicate regions.
Complementary to the CDK1-induced ubiquitination of the replisome, we have recently shown that the SUMO/ubiquitin equilibrium at active DNA replication forks also controls CDK1 activation. In 2016, we identified USP7 as a SUMO-dependent deubiquitinase that prevents excessive ubiquitination and SUMOylation in DNA replication forks [24,84]. Of note, the inhibition of USP7 induces the ubiquitination of many replication factors, including MCM7, at the canonical site responsible for replisome disassembly [24]. In parallel to the control of the ubiquitination of the replisome, USP7 also restricts the activity of CDK1 through the regulation of its phosphatase, PP2A [139]. As a consequence, blocking USP7 reduces PP2A activity and leads to the activation of CDK1. Concomitantly, an increase in the ubiquitination of the replisome results in premature disassembly of the replication machinery and generation of CDK1-dependent DNA damage in the S phase. Thus, the ubiquitin system constitutes an additional layer of regulation during cell cycle progression that connects the termination of DNA replication and the activation of CDK1 through regulated disassembly of the replication machinery. Beyond the control imposed by the accumulation of CDK activity, the ubiquitin–CDK1 relay could explain, at least in part, how active DNA replication prevents premature activation of the mitotic program. At the same time, this model provides a mechanism of how the unloading of the replication machinery is stimulated during the late S phase and mitosis to avoid problems after cell division. Whether the group SUMOylation of the replisome also plays a role in setting the stage for the ubiquitination of the CMG remains to be determined.
In addition to helping us understand how cells coordinate the completion of DNA replication with activation of the mitotic program, the ubiquitin–CDK1 connection has important clinical implications, since many inhibitors in this pathway are currently being studied as anticancer agents. We propose that the activation of CDK1 is an attractive therapeutic avenue, since it will induce DNA damage during DNA replication in combination with premature entry into mitosis that can lead to mitotic catastrophe and cell death (Figure 3B). A similar effect has been observed with ATR inhibitors that combine the generation of damage with the loss of the G2/M checkpoint [140,141]. Working through a different mechanism, the inhibition of USP7 can also elicit CDK1-dependent DNA damage and premature activation of the mitotic program. In this sense, we have shown that CDK1 is essential for the toxic effects of USP7 inhibitors in cancer cells. In addition, mutations that influence the status of CDK1 in cancer cells will be determined for the application of USP7 inhibitors in the clinic.
Author Contributions
A.G. and P.V. wrote the manuscript and prepared the figures. O.F.-C. and E.L. designed and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by two grants from MINECO (BFU2014-55168-JIN; RTI2018-093485-B-I00) and a Ramón y Cajal Fellowship from MINECO (RYC-2016-20705), co-funded by European Regional Development Funds (FEDER) to Emilio Lecona; by grants from the Spanish Ministry of Science, Innovation and Universities (RTI2018-102204-B-I00, co-financed with European FEDER funds) and the European Research Council (ERC-617840) to Oscar Fernandez-Capetillo; fellowships from Fundacion Ramon Areces-UAM and LaCaixa Foundation to Pablo Valledor (LCF/BQ/ES18/11670008) and a PhD fellowship from MINECO to Antonio Galarreta (BES-2015-075758).
Conflicts of Interest
The authors declare no competing financial interests.
References
- Damgaard, R.B. The ubiquitin system: From cell signalling to disease biology and new therapeutic opportunities. Cell Death Differ. 2021, 28, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Meluh, P.B.; Koshland, D. Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol. Biol. Cell 1995, 6, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Seufert, W.; Futcher, B.; Jentsch, S. Role of a ubiquitin-conjugating enzyme in degradation of S- and M-phase cyclins. Nature 1995, 373, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.P.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric Chains of SUMO-2 and SUMO-3 are Conjugated to Protein Substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, A.; Ackermann, L.; Hoppe, T. Ring of change: CDC48/p97 drives protein dynamics at chromatin. Front. Genet. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef]
- Vaz, B.; Halder, S.; Ramadan, K. Role of p97/VCP (Cdc48) in genome stability. Front. Genet. 2013, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y. Diverse functions with a common regulator: Ubiquitin takes command of an AAA ATPase. J. Struct. Biol. 2006, 156, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Dai, R.M.; Li, C.-C.H. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol. 2001, 3, 740–744. [Google Scholar] [CrossRef]
- Meyer, H.H.; Shorter, J.G.; Seemann, J.; Pappin, D.; Warren, G. A complex of mammalian Ufd1 and Npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000, 19, 2181–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik, C.; Yano, M.; DeMartino, G.N. RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J. Cell Sci. 2004, 117, 281–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, W.; Verma, R.; Kleiger, G.; Oania, R.S.; Deshaies, R.J. NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat. Struct. Mol. Biol. 2012, 19, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Bergink, S.; Ammon, T.; Kern, M.; Schermelleh, L.; Leonhardt, H.; Jentsch, S. Role of Cdc48/p97 as a SUMO-targeted segregase curbing Rad51-Rad52 interaction. Nat. Cell Biol. 2013, 15, 526–532. [Google Scholar] [CrossRef]
- Køhler, J.B.; Tammsalu, T.; Jørgensen, M.M.; Steen, N.; Hay, R.T.; Thon, G. Targeting of SUMO substrates to a Cdc48-Ufd1-Npl4 segregase and STUbL pathway in fission yeast. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Aslanian, A.; Prudden, J.; Heideker, J.; Vashisht, A.A.; Wohlschlegel, J.A.; Yates, J.R.; Boddy, M.N. Dual recruitment of Cdc48 (p97)-Ufd1-Npl4 ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO) and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stability functions. J. Biol. Chem. 2012, 287, 29610–29619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.H.; Wang, Y.; Warren, G. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 2002, 21, 5645–5652. [Google Scholar] [CrossRef]
- Elsasser, S.; Finley, D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 2005, 7, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.M.; Brasseur, C.; Meyer, H.H. The AAA ATPase p97/VCP interacts with its alternative co-factors, Ufd1-Np14 and p47, through a common bipartite binding mechanism. J. Biol. Chem. 2004, 279, 49609–49616. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, P.; Buchberger, A.; Schindelin, H. Hierarchical binding of cofactors to the AAA ATPase p97. Structure 2011, 19, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef]
- Kunz, K.; Piller, T.; Müller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Keiten-Schmitz, J.; Schunck, K.; Müller, S. SUMO Chains Rule on Chromatin Occupancy. Front. Cell Dev. Biol. 2020, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lecona, E.; Rodriguez-Acebes, S.; Specks, J.; Lopez-Contreras, A.J.; Ruppen, I.; Murga, M.; Muñoz, J.; Mendez, J.; Fernandez-Capetillo, O. USP7 is a SUMO deubiquitinase essential for DNA replication. Nat. Struct. Mol. Biol. 2016, 23, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Schimmel, J.; Eifler, K.; Olsen, J.V.; Vertegaal, A.C.O. Ubiquitin-specific Protease 11 (USP11) Deubiquitinates Hybrid Small Ubiquitin-like Modifier (SUMO)-Ubiquitin Chains to Counteract RING Finger Protein 4 (RNF4). J. Biol. Chem. 2015, 290, 15526–15537. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Boddy, M.N. Cooperativity of the SUMO and ubiquitin pathways in genome stability. Biomolecules 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.; Nassar, J.; Peiffer, I.; Stanojcic, S.; Sterkers, Y.; Delamarre, A.; Bocquet, S.; Méchali, M. The ORC ubiquitin ligase OBI1 promotes DNA replication origin firing. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Méndez, J.; Zou-Yang, X.H.; Kim, S.Y.; Hidaka, M.; Tansey, W.P.; Stillman, B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol. Cell 2002, 9, 481–491. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Q.; Liao, R.; Sun, P.; Wu, X. The SCF(Skp2) Ubiquitin Ligase Complex Interacts with the Human Replication Licensing Factor Cdt1 and Regulates Cdt1 Degradation*. J. Biol. Chem. 2003, 278, 30854–30858. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Pérez, S.; Cabrera, E.; Amoedo, H.; Rodríguez-Acebes, S.; Koundrioukoff, S.; Debatisse, M.; Méndez, J.; Freire, R. USP37 deubiquitinates Cdt1 and contributes to regulate DNA replication. Mol. Oncol. 2016, 10, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Nishitani, H.; Sugimoto, N.; Roukos, V.; Nakanishi, Y.; Saijo, M.; Obuse, C.; Tsurimoto, T.; Nakayama, K.I.; Nakayama, K.; Fujita, M.; et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006, 25, 1126–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Blow, J.J. Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA re-replication in Xenopus. EMBO J. 2005, 24, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, N.; Kitabayashi, I.; Osano, S.; Tatsumi, Y.; Yugawa, T.; Narisawa-Saito, M.; Matsukage, A.; Kiyono, T.; Fujita, M. Identification of Novel Human Cdt1-binding Proteins by a Proteomics Approach: Proteolytic Regulation by APC/CCdh1. Mol. Biol. Cell 2008, 19, 1007–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, E.E.; Walter, J.C. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 2006, 8, 84–90. [Google Scholar] [CrossRef]
- Zhong, W.; Feng, H.; Santiago, F.E.; Kipreos, E.T. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature 2003, 423, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Ralph, E.; Boye, E.; Kearsey, S.E. DNA damage induces Cdt1 proteolysis in fission yeast through a pathway dependent on Cdt2 and Ddb1. EMBO Rep. 2006, 7, 1134–1139. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kipreos, E.T. The Caenorhabditis elegans Replication Licensing Factor CDT-1 Is Targeted for Degradation by the CUL-4/DDB-1 Complex. Mol. Cell. Biol. 2007, 27, 1394–1406. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; McCall, C.M.; Ohta, T.; Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 2004, 6, 1003–1009. [Google Scholar] [CrossRef]
- Senga, T.; Sivaprasad, U.; Zhu, W.; Jong, J.P.; Arias, E.E.; Walter, J.C.; Dutta, A. PCNA is a cofactor for CDT1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 2006, 281, 6246–6252. [Google Scholar] [CrossRef] [Green Version]
- Sansam, C.L.; Shepard, J.L.; Lai, K.; Ianari, A.; Danielian, P.S.; Amsterdam, A.; Hopkins, N.; Lees, J.A. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 2006, 20, 3117–3129. [Google Scholar] [CrossRef] [Green Version]
- Higa, L.A.; Banks, D.; Wu, M.; Kobayashi, R.; Sun, H.; Zhang, H. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 2006, 5, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.O.; Wagener, C.; Marinoni, F.; Kramer, E.R.; Melixetian, M.; Denchi, E.L.; Gieffers, C.; Matteucci, C.; Peters, J.M.; Helin, K. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 2000, 14, 2330–2343. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Diffley, J.F.X. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005, 122, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, D.; Hoffmann, S.; Komseli, E.S.; Rappsilber, J.; Gorgoulis, V.; Sørensen, C.S. SCF(Cyclin F)-dependent degradation of CDC6 suppresses DNA re-replication. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, M.; Calzada, A.; Bueno, A. The Cdc6 protein is ubiquitinated in vivo for proteolysis in Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 9092–9097. [Google Scholar] [CrossRef] [Green Version]
- Perkins, G.; Drury, L.S.; Diffley, J.F.X. Separate SCF(CDC4) recognition elements target Cdc6 for proteolysis in S phase and mitosis. EMBO J. 2001, 20, 4836–4845. [Google Scholar] [CrossRef] [Green Version]
- Drury, L.S.; Perkins, G.; Diffley, J.F.X. The Cdc4/34/53 pathway targets Cdc6p for proteolysis in budding yeast. EMBO J. 1997, 16, 5966–5976. [Google Scholar] [CrossRef] [Green Version]
- Ballabeni, A.; Melixetian, M.; Zamponi, R.; Masiero, L.; Marinoni, F.; Helin, K. Human geminin promotes pre-RC formation and DNA replication by stabilizing CDT1 in mitosis. EMBO J. 2004, 23, 3122–3132. [Google Scholar] [CrossRef] [Green Version]
- McGarry, T.J.; Kirschner, M.W. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Pérez, S.; Cabrera, E.; Salido, E.; Lim, M.; Reid, L.; Lakhani, S.R.; Khanna, K.K.; Saunus, J.M.; Freire, R. DUB3 and USP7 de-ubiquitinating enzymes control replication inhibitor Geminin: Molecular characterization and associations with breast cancer. Oncogene 2017, 36, 4802–4809. [Google Scholar] [CrossRef] [PubMed]
- Charrasse, S.; Gharbi-Ayachi, A.; Burgess, A.; Vera, J.; Hached, K.; Raynaud, P.; Schwob, E.; Lorca, T.; Castro, A. Ensa controls S-phase length by modulating Treslin levels. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Pollok, S.; Grosse, F. Cdc45 degradation during differentiation and apoptosis. Biochem. Biophys. Res. Commun. 2007, 362, 910–915. [Google Scholar] [CrossRef]
- Bassermann, F.; Frescas, D.; Guardavaccaro, D.; Busino, L.; Peschiaroli, A.; Pagano, M. The Cdc14B-Cdh1-Plk1 Axis Controls the G2 DNA-Damage-Response Checkpoint. Cell 2008, 134, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Martín, Y.; Cabrera, E.; Amoedo, H.; Hernández-Pérez, S.; Domínguez-Kelly, R.; Freire, R. USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene 2015, 34, 1058–1063. [Google Scholar] [CrossRef]
- McGarry, E.; Gaboriau, D.; Rainey, M.D.; Restuccia, U.; Bachi, A.; Santocanale, C. The deubiquitinase USP9X maintains DNA replication fork stability and DNA damage checkpoint responses by regulating CLASPIN during S-PhaseH. Cancer Res. 2016, 76, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Bekker-Jensen, S.; Bartek, J.; Lukas, J. Destruction of Claspin by SCFβTrCP Restrains Chk1 Activation and Facilitates Recovery from Genotoxic Stress. Mol. Cell 2006, 23, 307–318. [Google Scholar] [CrossRef]
- Peschiaroli, A.; Dorrello, N.V.; Guardavaccaro, D.; Venere, M.; Halazonetis, T.; Sherman, N.E.; Pagano, M. SCFβTrCP-Mediated Degradation of Claspin Regulates Recovery from the DNA Replication Checkpoint Response. Mol. Cell 2006, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mamely, I.; van Vugt, M.A.; Smits, V.A.; Semple, J.I.; Lemmens, B.; Perrakis, A.; Medema, R.H.; Freire, R. Polo-like Kinase-1 Controls Proteasome-Dependent Degradation of Claspin during Checkpoint Recovery. Curr. Biol. 2006, 16, 1950–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustrup, H.; Bekker-Jensen, S.; Bartek, J.; Lukas, J.; Mailand, N. USP7 counteracts SCFβTrCP-but not APCCdh1-mediated proteolysis of Claspin. J. Cell Biol. 2009, 184, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Collyer, T.; Hardy, C.F. Cell cycle regulation of DNA replication Initiator Factor Dbf4p. Mol. Cell. Biol. 1999, 19, 4270–4278. [Google Scholar] [CrossRef] [Green Version]
- Godinho Ferreira, M.; Santocanale, C.; Drury, L.S.; Diffley, J.F.X. Dbf4p, an Essential S Phase-Promoting Factor, Is Targeted for Degradation by the Anaphase-Promoting Complex. Mol. Cell. Biol. 2000, 20, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Weinreich, M.; Stillman, B. Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J. 1999, 18, 5334–5346. [Google Scholar] [CrossRef]
- Psakhye, I.; Castellucci, F.; Branzei, D. SUMO-Chain-Regulated Proteasomal Degradation Timing Exemplified in DNA Replication Initiation. Mol. Cell 2019, 76, 632–645.e6. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Wei, L.; Peng, X.P.; Zhao, X. Sumoylation of the DNA polymerase ε by the Smc5/6 complex contributes to DNA replication. PLoS Genet. 2019, 15, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Winczura, A.; Appanah, R.; Tatham, M.H.; Hay, R.T.; De Piccoli, G. The S phase checkpoint promotes the Smc5/6 complex dependent SUMOylation of Pol2, the catalytic subunit of DNA polymerase ε. PLoS Genet. 2019, 15, e1008427. [Google Scholar] [CrossRef]
- Wei, L.; Zhao, X. A new MCM modification cycle regulates DNA replication initiation. Nat. Struct. Mol. Biol. 2016, 23, 209–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Albuquerque, C.P.; Liang, J.; Gaut, N.J.; Zhou, H. Molecular circuitry of the SUMO (Small Ubiquitin-like Modifier) pathway in controlling sumoylation homeostasis and suppressing genome rearrangements. J. Biol. Chem. 2016, 291, 8825–8835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maric, M.; Maculins, T.; De Piccoli, G.; Labib, K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 2014, 346. [Google Scholar] [CrossRef] [Green Version]
- Villa, F.; Fujisawa, R.; Ainsworth, J.; Nishimura, K.; Lie-a-ling, M.; Lacaud, G.; Labib, K.P.M. CUL2LRR1, TRAIP and p97 control CMG helicase disassembly in the mammalian cell cycle. EMBO J. 2021, 22, 1–13. [Google Scholar] [CrossRef]
- Sonneville, R.; Moreno, S.P.; Knebel, A.; Johnson, C.; Hastie, C.J.; Gartner, A.; Gambus, A.; Labib, K. CUL-2LRR-1 and UBXN-3 drive replisome disassembly during DNA replication termination and mitosis. Nat. Cell Biol. 2017, 19, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Dewar, J.M.; Low, E.; Mann, M.; Räschle, M.; Walter, J.C. CRL2Lrr1 promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev. 2017, 31, 275–290. [Google Scholar] [CrossRef] [Green Version]
- Vrtis, K.B.; Dewar, J.M.; Chistol, G.; Wu, R.A.; Graham, T.G.W.; Walter, J.C. Single-strand DNA breaks cause replisome disassembly. Mol. Cell 2021, 81, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wu, R.A.; Sonneville, R.; Kochenova, O.V.; Labib, K.; Pellman, D.; Walter, J.C. Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol. Cell 2019, 73, 915–929.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.P.; Jones, R.M.; Poovathumkadavil, D.; Scaramuzza, S.; Gambus, A. Mitotic replisome disassembly depends on TRAIP ubiquitin ligase activity. Life Sci. Alliance 2019, 2, 1–12. [Google Scholar] [CrossRef]
- Wu, R.A.; Semlow, D.R.; Kamimae-Lanning, A.N.; Kochenova, O.V.; Chistol, G.; Hodskinson, M.R.; Amunugama, R.; Sparks, J.L.; Wang, M.; Deng, L.; et al. TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 2019, 567, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Diffley, J.F.X. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat. Cell Biol. 2002, 4, 198–207. [Google Scholar] [CrossRef]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef]
- Tanaka, S.; Umemori, T.; Hirai, K.; Muramatsu, S.; Kamimura, Y.; Araki, H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 2007, 445, 328–332. [Google Scholar] [CrossRef]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic Origin-Dependent DNA Replication In Vitro Reveals Sequential Action of DDK and S-CDK Kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Ilves, I.; Petojevic, T.; Pesavento, J.J.; Botchan, M.R. Activation of the MCM2-7 Helicase by Association with Cdc45 and GINS Proteins. Mol. Cell 2010, 37, 247–258. [Google Scholar] [CrossRef]
- Lopez-Contreras, A.J.; Ruppen, I.; Nieto-Soler, M.; Murga, M.; Rodriguez-Acebes, S.; Remeseiro, S.; Rodrigo-Perez, S.; Rojas, A.M.; Mendez, J.; Muñoz, J.; et al. A Proteomic Characterization of Factors Enriched at Nascent DNA Molecules. Cell Rep. 2013, 3, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Alabert, C.; Bukowski-Wills, J.C.; Lee, S.B.; Kustatscher, G.; Nakamura, K.; De Lima Alves, F.; Menard, P.; Mejlvang, J.; Rappsilber, J.; Groth, A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014, 16, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. A SUMO and ubiquitin code coordinates protein traffic at replication factories. BioEssays 2016, 38, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.P.; Labib, K. Chromosome duplication in Saccharomyces cerevisiae. Genetics 2016, 203, 1027–1067. [Google Scholar] [CrossRef] [Green Version]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA replication fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef]
- Deegan, T.D.; Diffley, J.F.X. MCM: One ring to rule them all. Curr. Opin. Struct. Biol. 2016, 37, 145–151. [Google Scholar] [CrossRef]
- Dewar, J.M.; Walter, J.C. Mechanisms of DNA replication termination. Nat. Rev. Mol. Cell Biol. 2017, 18, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.P.; Gambus, A. Mechanisms of eukaryotic replisome disassembly. Biochem. Soc. Trans. 2020, 48, 823–836. [Google Scholar] [CrossRef]
- Gambus, A. Termination of eukaryotic replication forks. Adv. Exp. Med. Biol. 2017, 1042, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, A.; Minchell, N.E.; Baxter, J. The causes and consequences of topological stress during DNA replication. Genes 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Been, D.M.; Champoux, J.J. Breakage of single-stranded DNA by rat liver nicking-closing enzyme with the formation of a DNA-enzyme complex. Nucleic Acids Res. 1980, 8, 6129–6142. [Google Scholar] [CrossRef] [Green Version]
- Deegan, T.D.; Baxter, J.; Ortiz Bazán, M.Á.; Yeeles, J.T.P.; Labib, K.P.M. Pif1-Family Helicases Support Fork Convergence during DNA Replication Termination in Eukaryotes. Mol. Cell 2019, 74, 231–244.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espeli, O.; Levine, C.; Hassing, H.; Marians, K.J. Temporal regulation of topoisomerase IV activity in E. coli. Mol. Cell 2003, 11, 189–201. [Google Scholar] [CrossRef]
- Heintzman, D.R.; Campos, L.V.; Byl, J.A.W.; Osheroff, N.; Dewar, J.M. Topoisomerase II Is Crucial for Fork Convergence during Vertebrate Replication Termination. Cell Rep. 2019, 29, 422–436.e5. [Google Scholar] [CrossRef] [Green Version]
- Hiasa, H.; Marians, K.J. Two distinct modes of strand unlinking during θ-type DNA replication. J. Biol. Chem. 1996, 271, 21529–21535. [Google Scholar] [CrossRef] [Green Version]
- Ishimi, Y.; Sugasawa, K.; Hanaoka, F.; Eki, T.; Hurwitz, J. Topoisomerase II plays an essential role as a swivelase in the late stage of SV40 chromosome replication in vitro. J. Biol. Chem. 1992, 267, 462–466. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.Y.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Postow, L.; Crisona, N.J.; Peter, B.J.; Hardy, C.D.; Cozzarelli, N.R. Topological challenges to DNA replication: Conformations at the fork. Proc. Natl. Acad. Sci. USA 2001, 98, 8219–8226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Dewar, J.M.; Budzowska, M.; Walter, J.C. The mechanism of DNA replication termination in vertebrates. Nature 2015, 525, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Lucas, I.; Germe, T.; Chevrier-Miller, M.; Hyrien, O. Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J. 2001, 20, 6509–6519. [Google Scholar] [CrossRef] [Green Version]
- Zechiedrich, E.L.; Cozzarelli, N.R. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 11801–11805. [Google Scholar] [CrossRef] [Green Version]
- Labib, K.; Tercero, J.A.; Diffley, J.F.X. Uninterrupted MCMZ-7 Function Required for DNA Replication Fork Progression. Science 2000, 288, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Mukherjee, P.; Tatham, M.H.; Hay, R.; Labib, K. Ufd1-Npl4 Recruit Cdc48 for Disassembly of Ubiquitylated CMG Helicase at the End of Chromosome Replication. Cell Rep. 2017, 18, 3033–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, A.; Orth, M.; Pirson, P.A.; Sonneville, R.; Blow, J.J.; Gartner, A.; Stemmann, O.; Hoppe, T. CDC-48/p97 Coordinates CDT-1 Degradation with GINS Chromatin Dissociation to Ensure Faithful DNA Replication. Mol. Cell 2011, 44, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.P.; Bailey, R.; Campion, N.; Herron, S.; Gambus, A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 2014, 346, 477–481. [Google Scholar] [CrossRef]
- Low, E.; Chistol, G.; Zaher, M.S.; Kochenova, O.V.; Walter, J.C. The DNA replication fork suppresses CMG unloading from chromatin before termination. Genes Dev. 2020, 34, 1534–1545. [Google Scholar] [CrossRef]
- Deegan, T.D.; Mukherjee, P.P.; Fujisawa, R.; Rivera, C.P.; Labib, K. Cmg helicase disassembly is controlled by replication fork DNA, replisome components and a ubiquitin threshold. Elife 2020, 9, 1–33. [Google Scholar] [CrossRef]
- Maculins, T.; Nkosi, P.J.; Nishikawa, H.; Labib, K. Tethering of SCFDia2 to the Replisome Promotes Efficient Ubiquitylation and Disassembly of the CMG Helicase. Curr. Biol. 2015, 25, 2254–2259. [Google Scholar] [CrossRef] [Green Version]
- Mimura, S.; Komata, M.; Kishi, T.; Shirahige, K.; Kamura, T. SCFDia2 regulates DNA replication forks during S-phase in budding yeast. EMBO J. 2009, 28, 3693–3705. [Google Scholar] [CrossRef] [Green Version]
- Morohashi, H.; Maculins, T.; Labib, K. The Amino-Terminal TPR Domain of Dia2 Tethers SCFDia2 to the Replisome Progression Complex. Curr. Biol. 2009, 19, 1943–1949. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.P.; Labib, K.P.M. In Vitro Reconstitution Defines the Minimal Requirements for Cdc48-Dependent Disassembly of the CMG Helicase in Budding Yeast. Cell Rep. 2019, 28, 2777–2783.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019, 365. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fujisawa, R.; Deegan, T.D.; Sonneville, R.; Labib, K.P.M. TIMELESS-TIPIN and UBXN-3 promote replisome disassembly during DNA replication termination in Caenorhabditis elegans. EMBO J. 2021, 1–18. [Google Scholar] [CrossRef]
- Jagannathan, M.; Nguyen, T.; Gallo, D.; Luthra, N.; Brown, G.W.; Saridakis, V.; Frappier, L. A Role for USP7 in DNA Replication. Mol. Cell. Biol. 2014, 34, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchberger, A.; Schindelin, H.; Hänzelmann, P. Control of p97 function by cofactor binding. FEBS Lett. 2015, 589, 2578–2589. [Google Scholar] [CrossRef] [Green Version]
- Fullbright, G.; Rycenga, H.B.; Gruber, J.D.; Long, D.T. p97 Promotes a Conserved Mechanism of Helicase Unloading during DNA Cross-Link Repair. Mol. Cell. Biol. 2016, 36, 2983–2994. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Smedegaard, S.; Nakamura, K.; Mortuza, G.B.; Räschle, M.; de Opakua, A.I.; Oka, Y.; Feng, Y.; Blanco, F.J.; Mann, M.; et al. TRAIP is a PCNA-binding ubiquitin ligase that protects genome stability after replication stress. J. Cell Biol. 2016, 212, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [Green Version]
- Burrow, A.A.; Williams, L.E.; Pierce, L.C.T.; Wang, Y.H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics 2009, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlt, M.F.; Durkin, S.G.; Ragland, R.L.; Glover, T.W. Common fragile sites as targets for chromosome rearrangements. DNA Repair 2006, 5, 1126–1135. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Lalai, A.S.; Hoeijmakers, J.H.J. Fanconi anemia (cross)linked to DNA repair. Cell 2005, 123, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of Disease Susceptibility Pathways in Fanconi’s Anemia and Breast Cancer. N. Engl. J. Med. 2010, 362, 1909–1928. [Google Scholar] [CrossRef] [Green Version]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Räschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of Replication-Coupled DNA Interstrand Crosslink Repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015, 22, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Long, D.T.; Joukov, V.; Budzowska, M.; Walter, J.C. BRCA1 promotes unloading of the CMG Helicase from a stalled DNA replication fork. Mol. Cell 2014, 56, 174–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semlow, D.R.; Zhang, J.; Budzowska, M.; Drohat, A.C.; Walter, J.C. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 2016, 167, 498–511.e14. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, J.; Wallace, S.S.; Chen, J.; Zhou, J.; D’Andrea, A.D. Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 3014–3028. [Google Scholar] [CrossRef]
- Akopyan, K.; Silva Cascales, H.; Hukasova, E.; Saurin, A.T.; Müllers, E.; Jaiswal, H.; Hollman, D.A.A.; Kops, G.J.P.L.; Medema, R.H.; Lindqvist, A. Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol. Cell 2014, 53, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 2018, 71, 117–128.e3. [Google Scholar] [CrossRef] [Green Version]
- Lemmens, B.; Lindqvist, A. DNA replication and mitotic entry: A brake model for cell cycle progression. J. Cell Biol. 2019, 218, 3892–3902. [Google Scholar] [CrossRef]
- Galarreta, A.; Valledor, P.; Ubieto-Capella, P.; Lafarga, V.; Zarzuela, E.; Muñoz, J.; Malumbres, M.; Lecona, E.; Fernandez-Capetillo, O. USP7 limits CDK1 activity throughout the cell cycle. EMBO J. 2021, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
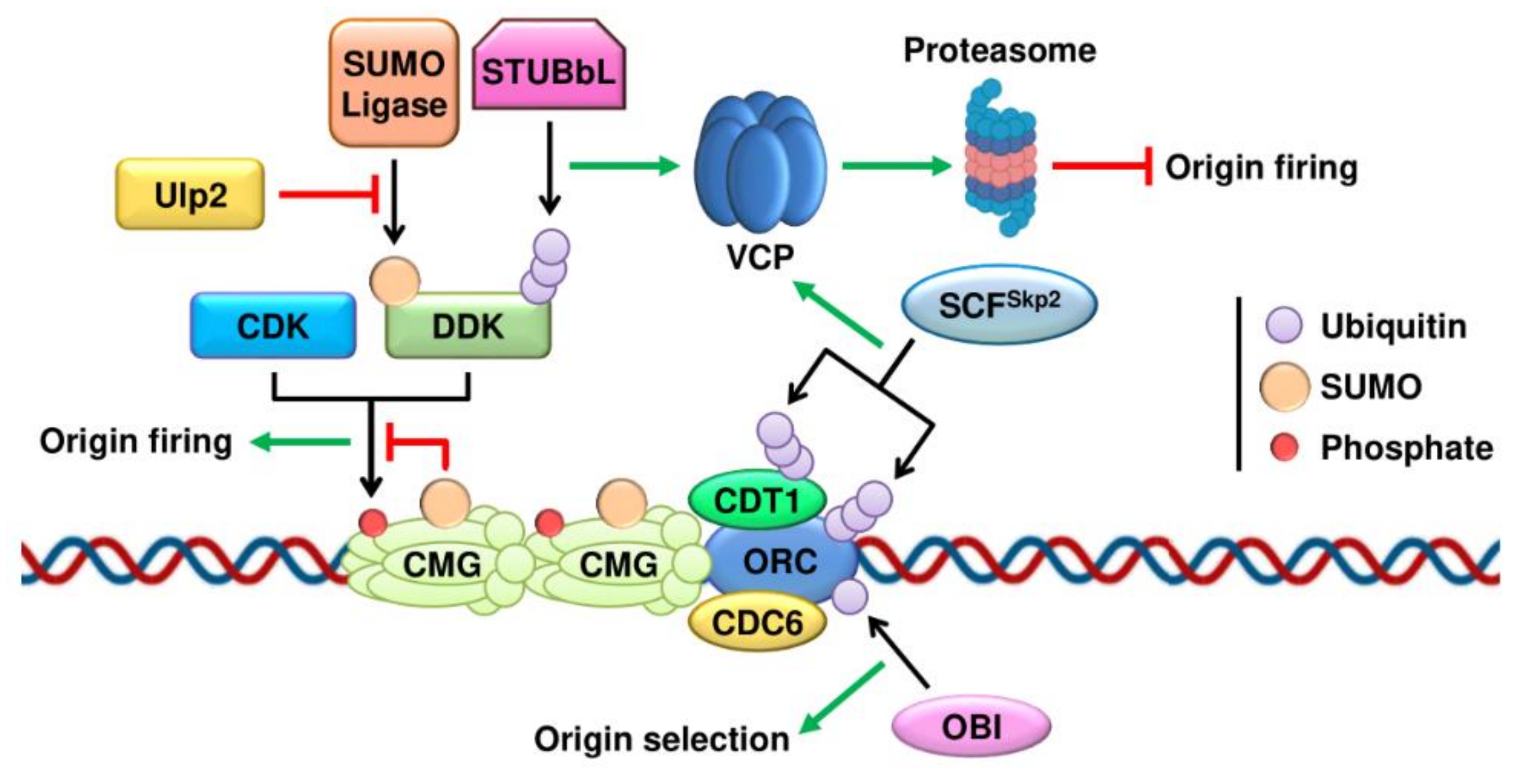
Figure 1.
Control of DNA replication initiation by the ubiquitin and SUMO pathways. Model for the assembly of the pre-RC complex and the different layers of control by ubiquitination and SUMOylation of the initiation machinery. CMG is the replicative helicase composed of CDC45, the MCM2-7 complex and the GINS complex.
Figure 1.
Control of DNA replication initiation by the ubiquitin and SUMO pathways. Model for the assembly of the pre-RC complex and the different layers of control by ubiquitination and SUMOylation of the initiation machinery. CMG is the replicative helicase composed of CDC45, the MCM2-7 complex and the GINS complex.

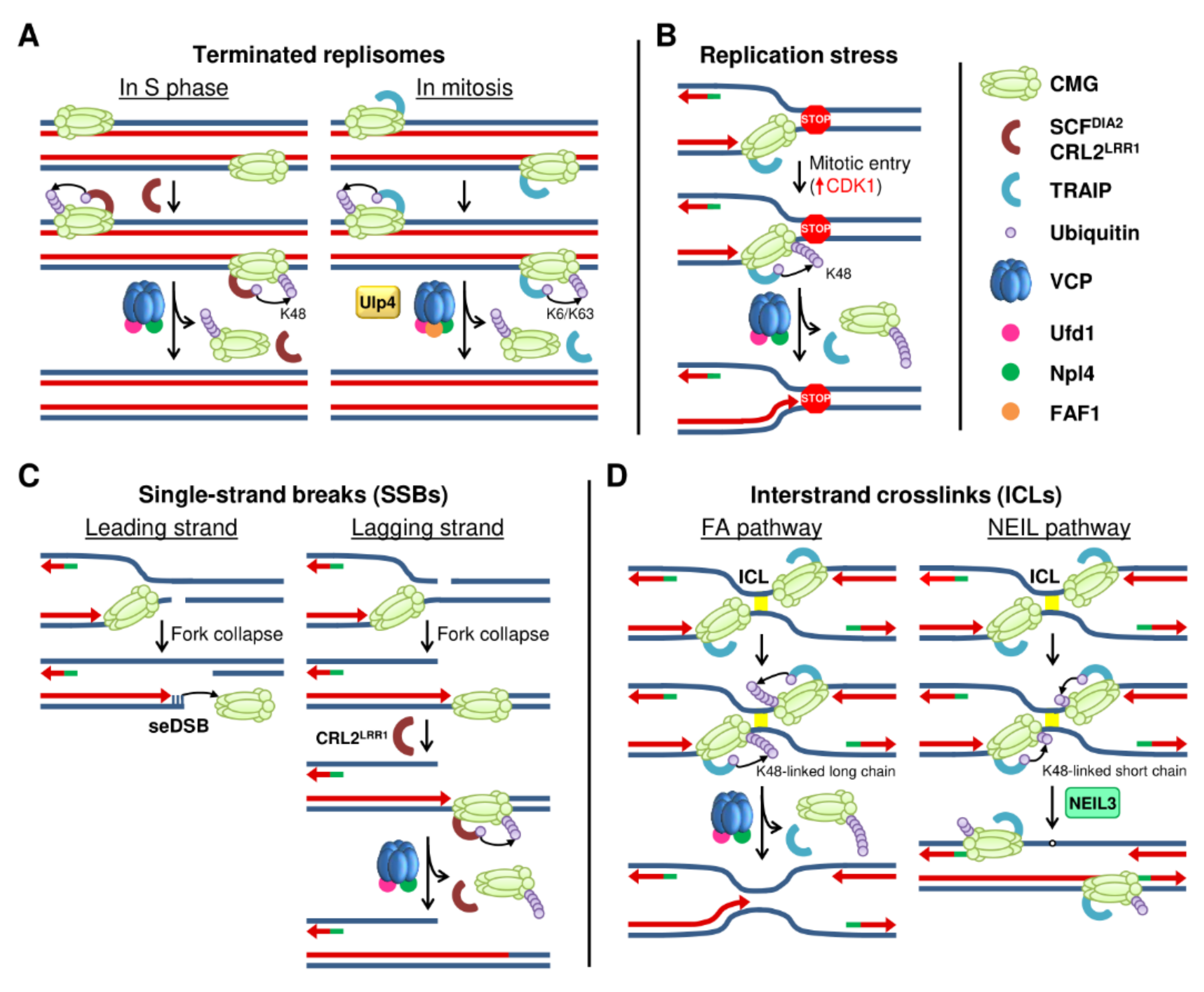
Figure 2.
Models for replisome disassembly. (A) Disassembly of terminated replisomes during the S phase (left) and in mitosis (right) is mediated by the ubiquitination of MCM7 with specific E3 ubiquitin ligases. (B) Replisome disassembly under replication stress is mediated by the CDK1-dependent activation of TRAIP to ubiquitinate MCM7. (C) Replisome disassembly in the presence of SSBs. When the SSB is in the leading strand (left), the CMG slides off the break, while a SSB in the lagging strand leads to CMG translocation along dsDNA and its ubiquitination by CRL2LRR1 (right). (D) In the presence of ICLs, replication forks convergence and MCM7 is ubiquitinated at K48 by TRAIP. Long ubiquitin chains lead to CMG unloading (left) and repair through the FA pathway, whereas short ubiquitin chains activate the NEIL3 glycosylase pathway (right).
Figure 2.
Models for replisome disassembly. (A) Disassembly of terminated replisomes during the S phase (left) and in mitosis (right) is mediated by the ubiquitination of MCM7 with specific E3 ubiquitin ligases. (B) Replisome disassembly under replication stress is mediated by the CDK1-dependent activation of TRAIP to ubiquitinate MCM7. (C) Replisome disassembly in the presence of SSBs. When the SSB is in the leading strand (left), the CMG slides off the break, while a SSB in the lagging strand leads to CMG translocation along dsDNA and its ubiquitination by CRL2LRR1 (right). (D) In the presence of ICLs, replication forks convergence and MCM7 is ubiquitinated at K48 by TRAIP. Long ubiquitin chains lead to CMG unloading (left) and repair through the FA pathway, whereas short ubiquitin chains activate the NEIL3 glycosylase pathway (right).

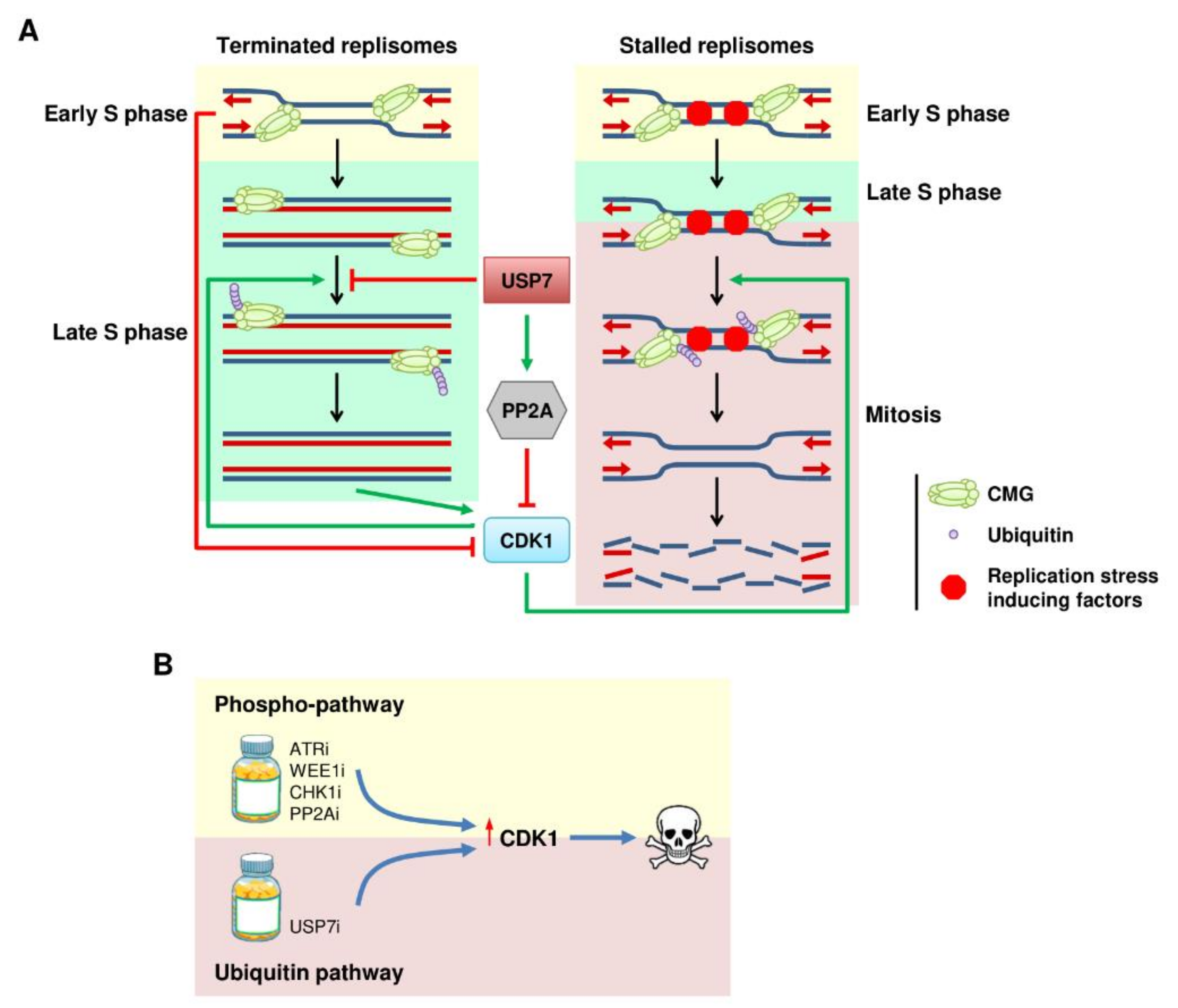
Figure 3.
Ubiquitination links DNA replication and CDK1. (A) Model for the ubiquitin/CDK1 relay in DNA replication. Active DNA replication forks and USP7 suppress CMG ubiquitination and CDK1 activation to prevent premature entry in mitosis. Accumulating CDK1 activity promotes CMG ubiquitination, leading to disassembly of the terminated replisomes before cell division, or disassembly of the stalled forks inducing DNA damage. (B) Therapeutic implications for the ubiquitin–CDK1 connection. Targeting the ubiquitin pathway might be an alternative way to prematurely activate CDK1 and lead to cell death.
Figure 3.
Ubiquitination links DNA replication and CDK1. (A) Model for the ubiquitin/CDK1 relay in DNA replication. Active DNA replication forks and USP7 suppress CMG ubiquitination and CDK1 activation to prevent premature entry in mitosis. Accumulating CDK1 activity promotes CMG ubiquitination, leading to disassembly of the terminated replisomes before cell division, or disassembly of the stalled forks inducing DNA damage. (B) Therapeutic implications for the ubiquitin–CDK1 connection. Targeting the ubiquitin pathway might be an alternative way to prematurely activate CDK1 and lead to cell death.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Regulation of DNA replication by Ub and SUMO.
| Target | Modification | E3 Ligase | Protease | Organism | Role | Cellular Context | References |
|---|---|---|---|---|---|---|---|
| ORC3/ORC5 | Ubiquitin | OBI1 | Human | Origin selection for firing | Replication initiation, G1/S | [27] | |
| ORC1 | Ubiquitin | SCFSkp2 | Human | Prevent re-licensing | Replication initiation, G1/S | [28] | |
| CDT1 | Ubiquitin | SCFSkp2 | USP37 | Human | Prevent re-licensing | Replication initiation, G1/S | [29,30,31] |
| APC/C-CDH1 | Human, Xenopus | Regulate licensing. Prevent licensing in quiescence. | M/G1 | [32,33] | |||
| CRL4-DDB1CDT2 | Human, yeast, Xenopus, C. elegans, zebrafish | Prevent re-licensing. Prevent licensing upon DNA damage | Replication initiation, G1/S | [31,34,35,36,37,38,39,40,41] | |||
| CDC6 (Cdc18 in yeast) | Ubiquitin | APC/C-CDH1 | Human | Prevent re-licensing. | Early G1 | [42] | |
| Prevent pre-RC assembly | Quiescence | [43] | |||||
| SCFCyclinF | Prevent re-licensing | G2 | [44] | ||||
| SCFCdc4 | Yeast | G1/S | [45,46,47] | ||||
| Geminin | Ubiquitin | APC/C-CDH1 | Dub3, USP7 | Xenopus | Promote origin licensing | M/G1 | [48,49,50] |
| Treslin | Ubiquitin | CRLs | Human | Prevent origin firing | G1/S | [51] | |
| CDC45 | Ubiquitin | APC/C-CDH1 | Human | Prevent origin firing | M/G1 | [52] | |
| Claspin | Ubiquitin | APC/C-CDH1 | USP28, USP29, USP9X | Human | Prevent origin firing. Recovery from G2 checkpoint response. | M/G1 | [53,54,55] |
| SCFβTrCP | USP7 | Human | Recovery from checkpoint response | G2/M | [56,57,58,59] | ||
| DBF4 | Ubiquitin | APC/C-Cdc20 | Yeast | Prevent re-replication. Prevent new pre-RC firing | M/G1 | [60,61,62] | |
| DDK | SUMO | Siz1, Siz2 | Ulp2 | Yeast | Prevent origin firing | Replication initiation | [63] |
| Ubiquitin | Slx5/Slx8 | Yeast | [63] | ||||
| Polymerase ε (Pol2 subunit) | SUMO | Smc5/6 complex (Mms21 subunit) | Yeast | Promote fork progression under replication stress | Elongation, S phase | [64,65] | |
| MCM7 | SUMO | Mms21, Siz1, Siz2 | Ulp2 | Yeast | Prevent origin firing | Replication initiation, G1 | [66,67] |
| Ubiquitin | SCFDia2 | Yeast | Trigger replisome disassembly | Replication termination | [68] | ||
| CRL2LRR1 | C. elegans, Xenopus, mouse embryonic stem cells | [69,70,71] | |||||
| Xenopus | Under lagging strand SSBs | [72] | |||||
| TRAIP | C. elegans, Xenopus, mouse embryonic stem cells | Trigger replisome disassembly | Mitosis | [69,73,74] | |||
| C. elegans, Xenopus | Trigger replisome disassembly | Stalled replisomes upon RS | [73] | ||||
| Xenopus | Trigger replisome disassembly | ICL repair by FA pathway | [75] | ||||
| NEIL3 recruitment | ICL repair by NEIL3 glycosylase pathway | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Galarreta, A.; Valledor, P.; Fernandez-Capetillo, O.; Lecona, E. Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1. Int. J. Mol. Sci. 2021, 22, 8796. https://doi.org/10.3390/ijms22168796
AMA Style
Galarreta A, Valledor P, Fernandez-Capetillo O, Lecona E. Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1. International Journal of Molecular Sciences. 2021; 22(16):8796. https://doi.org/10.3390/ijms22168796
Chicago/Turabian StyleGalarreta, Antonio, Pablo Valledor, Oscar Fernandez-Capetillo, and Emilio Lecona. 2021. "Coordinating DNA Replication and Mitosis through Ubiquitin/SUMO and CDK1" International Journal of Molecular Sciences 22, no. 16: 8796. https://doi.org/10.3390/ijms22168796
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.