
 EJCRIM CALL FOR DEPUTY EDITOR! view more details
EJCRIM CALL FOR DEPUTY EDITOR! view more details



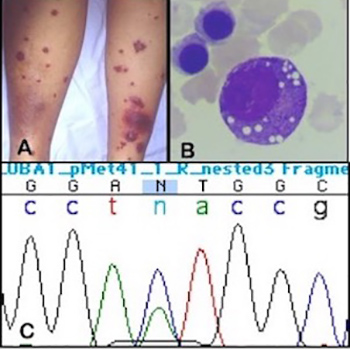
The VEXAS Syndrome: Uncontrolled Inflammation and Macrocytic Anaemia in a 77-Year-Old Male Patient
Andreas Himmelmann
 |
Hematology Practice, Lucerne, Switzerland |
Rolf Brücker
|
Rheumatology, Hirslanden Klinik St. Anna, Lucerne, Switzerland |
Keywords
Inflammatory syndromes, somatic mutation, macrocytic anemia
Abstract
The VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is a recently described X-linked autoinflammatory condition caused by a somatic mutation of the UBA1 gene and characterized by an evolving phenotype. This includes inflammatory processes such as recurrent fever, Sweet’s syndrome of the skin, pulmonary fibrosis, relapsing polychondritis and venous thromboembolism. An important feature, present in almost all cases, is the development of a macrocytic anaemia with vacuolization of myeloid and erythroid precursors. Usually, these patients require high doses of steroids to control symptoms and respond poorly to disease-modifying drugs.
We describe a new case of the VEXAS syndrome presenting with Sweet’s syndrome which has now been followed for 6 years.
References
- Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Eng J Med 2020;383(27):2628–2638.
- Levy-Lahad E, King M-C. Hiding in plain sight - somatic mutation in human disease. N Eng J Med 2020;383(27):2680–2682.
- Castaneda S, Martinez-Quintanilla D, Martin-Varillas JL, García-Castañeda N, Atienza-Mateo B, González-Gay MA. Tocilizumab for the treatment of adult-onset Still’s disease. Expert Opin Biol Ther 2019;19(4):273–286.
Views: 1870
HTML downloads: 776
PDF downloads: 1117
Published:
2021-04-13
Issue:
2021: Vol 8 No 4
(view)

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.







