Phosphodiesterase Type-5 Inhibitor Tadalafil Modulates Steroid Hormones Signaling in a Prostate Cancer Cell Line

 , , ,
, , ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
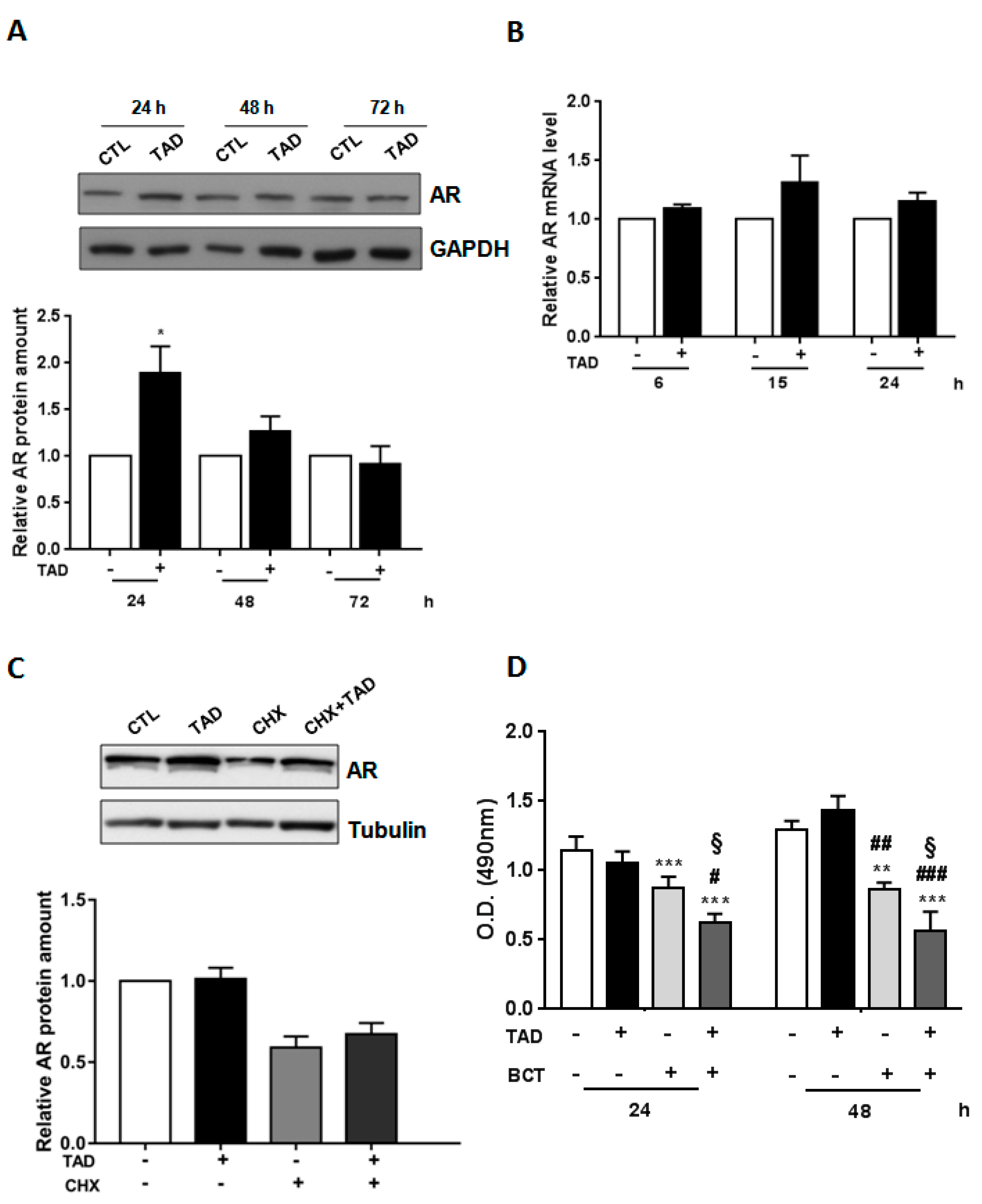
2.1. Tadalafil Increases AR Expression and Function without Affecting LNCaP Cell Viability and the Proliferation Rate
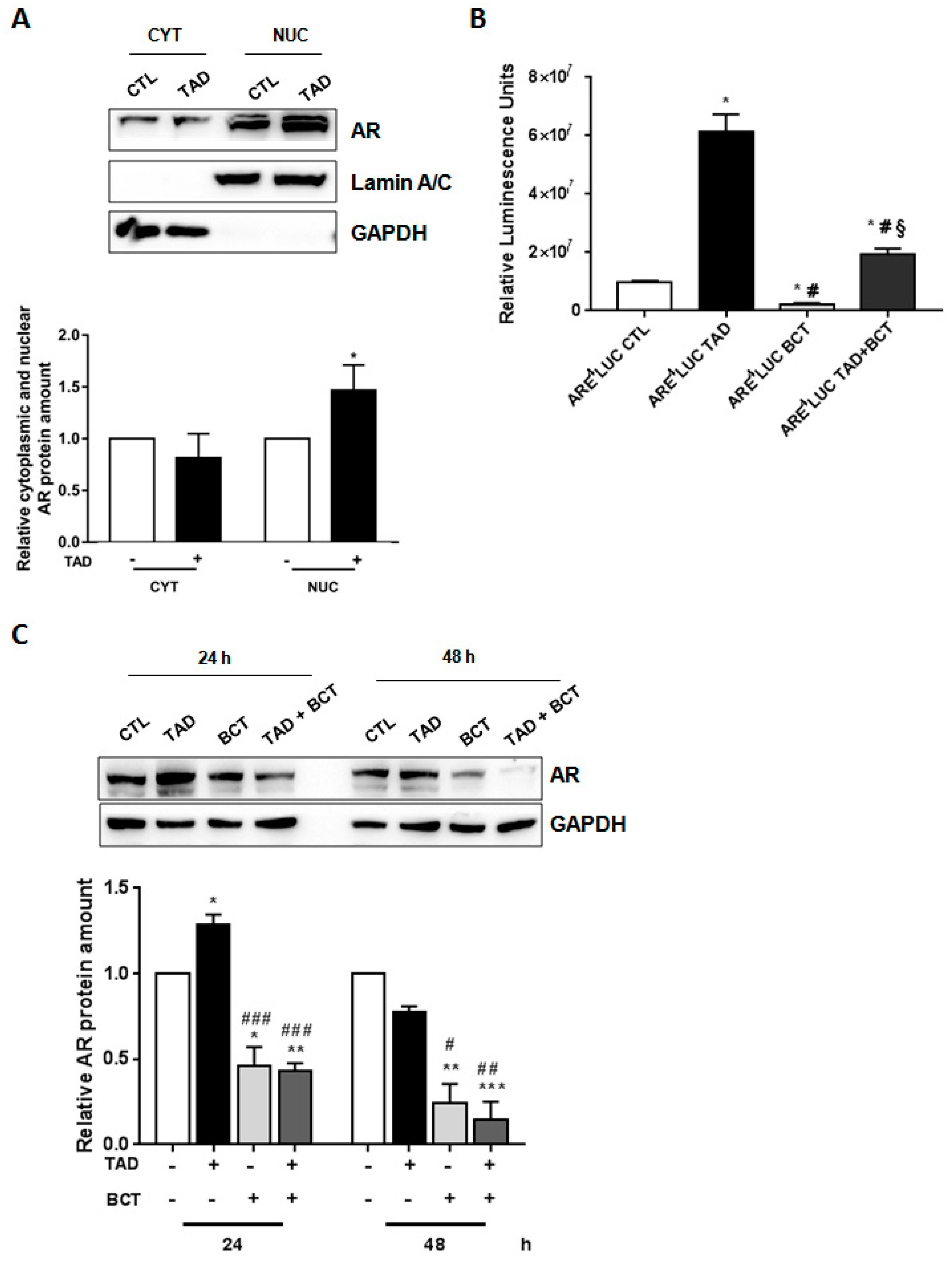
2.2. Tadalafil Promotes AR Nuclear Translocation and Transcriptional Activity
2.3. Tadalafil Increases In Vitro BCT-Mediated AR Downregulation and Cytostatic Effects on LNCaP Cells
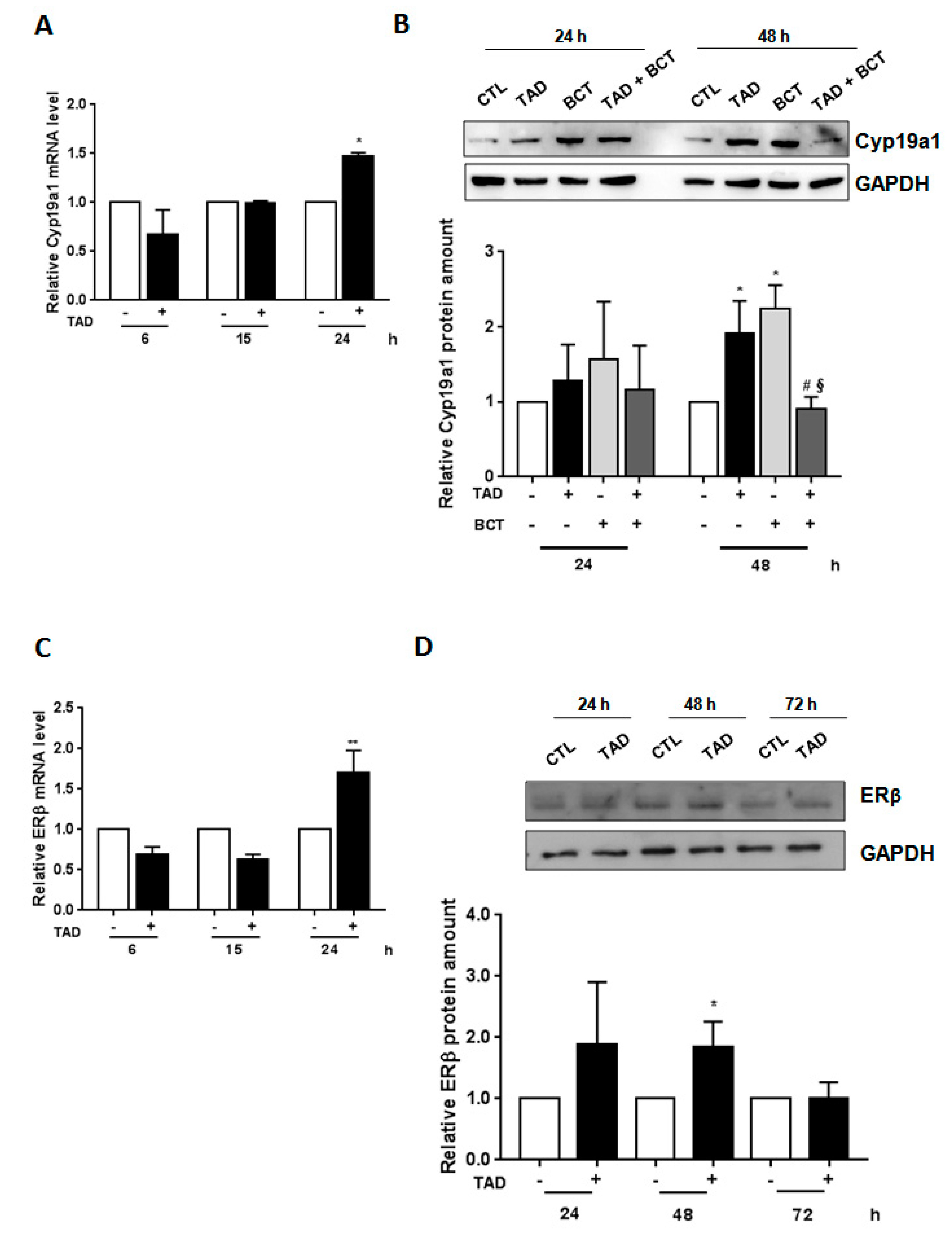
2.4. Tadalafil Modulates Aromatase and Estrogen Receptor-ß Expression
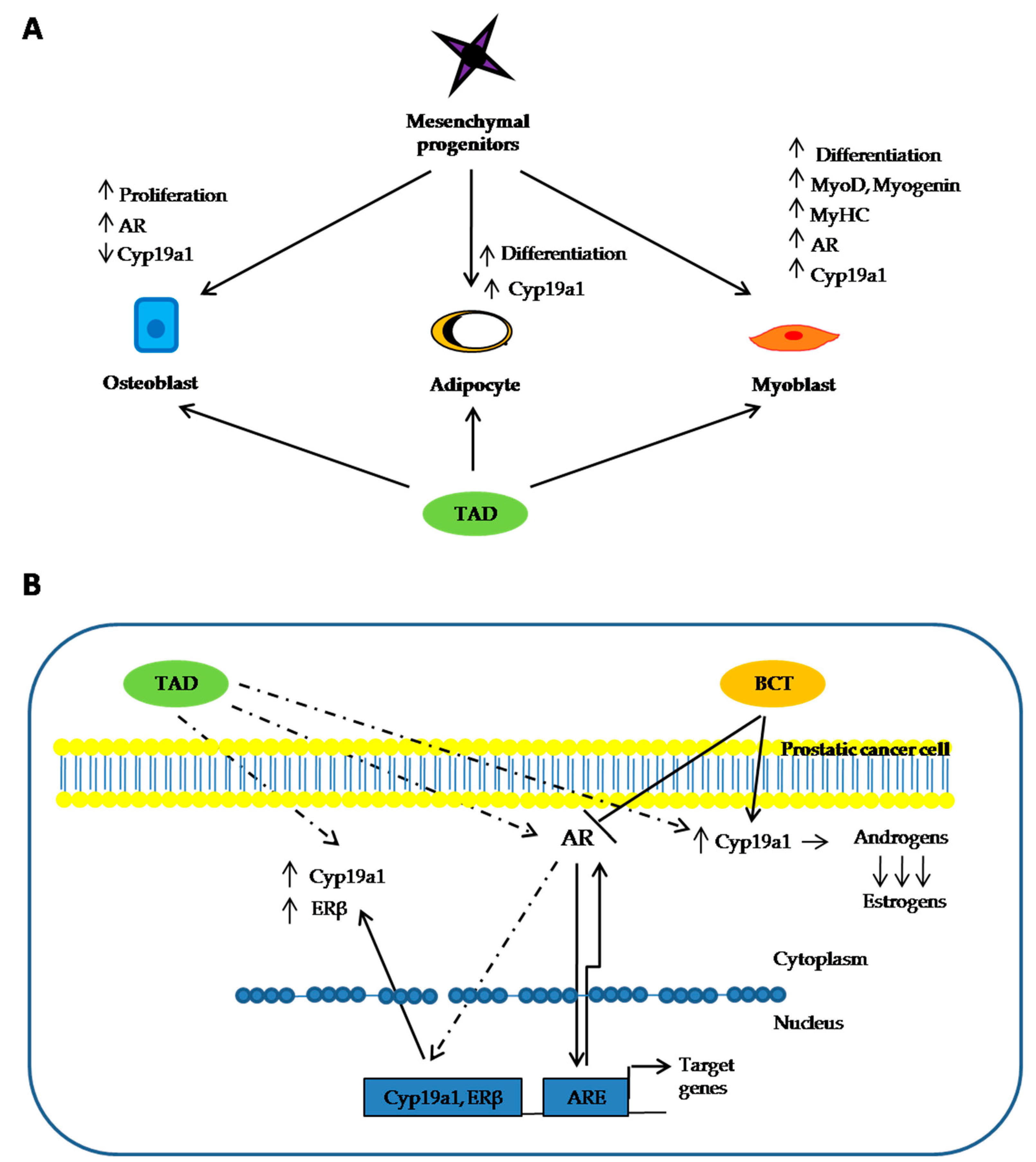
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. RNA Isolation and Quantitative Real-Time PCR
- -
- AR (NM_000044), FW: TAC CAG CTC ACC AAG CTC CT; REW: GAT GGG CTT GAC TTT CCC AG.
- -
- Cyp19a1 (NM_000103), FW: ACT ACA ACC GGG TAT ATG GAGAA; REW: TCG AGA GCT GTA ATG ATT GTGC.
- -
- ERβ (NM_001214902), FW: AGC ACG GCT CCA TAT ACA TACC; REW: TGG ACC ACT AAA GGA GAA AGGT
- -
- Cyclophilin A (NM_021130), FW: GTC AAC CCC ACC GTG TTC TT; REW: AAA GTT TTC TGC TGT TTT TGGAAT C.
- -
- PSA (NM_001030047), FW: GAGGAGTTCTTGACCCCAAAG; REW: CGCACACACGTCATTGGAAATA.
4.5. Protein Extraction and Western Blot Analysis
4.6. Luciferase Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AR | Androgen Receptor |
| PCa | Prostate cancer |
| Cyp19a1 | Aromatase |
| TAD | Tadalafil |
| LnCAP | Androgen-sensitive human PCa cell line |
| BCT | Bicalutamide |
| T | Testosterone |
| DHT | Dihydrotestosterone |
| AREs | Androgenr esponse elements |
| ADT | Androgen deprivation therapy |
| CRPC | Castration-resistant prostate cancer |
| cAMP | Cyclic adenosine monophosphate |
| cGMP | Cyclic guanosine monophosphate |
| PDE | Phosphodiesterase |
| PDE5i | PDE5 inhibitor |
| CHX | Cycloheximide |
| PSA | Prostatic specific antigen |
| MDV | MDV3100 |
| CYT | Cytoplasmic fraction |
| NUC | Nuclear fraction |
| RD | Human Rhabdomyosarcoma Cell Lines |
| ED | Erectile dysfunction |
| BDH | Benign prostatic hyperplasia |
| ERβ | Estrogen receptor beta |
| LUTS | Lower urinary tract symptoms |
| PSA | Prostatic specific antigen |
| PD-1 | Programmed cell death protein 1 |
| PD-1L | Programmed death-ligand 1 |
| AIs | Aromatase inhibitors |
| ERα | Estrogen receptor alpha |
| ERβ | Estrogen receptor beta |
| HGPIN | High grade prostatic intraepithelial neoplasia |
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancerstatistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a030452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, I.E.; Culig, Z.; Putz, T.; Nessler-Menardi, C.; Bartsch, G.; Klocker, H. Molecular biology of the androgen receptor: From molecular understanding to the clinic. Eur. Urol. 2001, 40, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2019, 22, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Wadosky, K.M.; Koochekpour, S. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget 2016, 7, 64447–64470. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. JAMA 2005, 294, 238–244. [Google Scholar] [CrossRef]
- Di Nunno, V.; Mollica, V.; Santoni, M.; Gatto, L.; Schiavina, R.; Fiorentino, M.; Brunocilla, E.; Ardizzoni, A.; Massari, F. New Hormonal Agents in Patients with Nonmetastatic Castration-Resistant Prostate Cancer: Meta-Analysis of Efficacy and Safety Outcomes. Clin. Genitourin. Cancer 2019, 17, e871–e877. [Google Scholar] [CrossRef]
- Albany, C.; Alva, A.S.; Aparicio, A.M.; Singal, R.; Yellapragada, S.; Sonpavde, G.; Hahn, N.M. Epigenetics in prostate cancer. Prostate Cancer 2011, 2011, 580318. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wu, L.Y.; Fulton, M.D.; Johnson, J.M.; Berkman, C.E. Prolonged androgen deprivation leads to downregulation of androgen receptor and prostate-specific membrane antigen in prostate cancer cells. Int. J. Oncol. 2012, 41, 2087–2092. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Sawyers, C.L. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 2005, 23, 8253–8261. [Google Scholar] [CrossRef]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phophodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Levy, I.; Horvath, A.; Azevedo, M.; de Alexandre, R.B.; Stratakis, C.A. Phosphodiesterase function and endocrine cells: Links to human disease and roles in tumor development and treatment. Curr. Opin. Pharmacol. 2011, 11, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Catalano, S.; Campana, A.; Giordano, C.; Gyorffy, B.; Tarallo, R.; Rinaldi, A.; Bruno, G.; Ferraro, A.; Romeo, F.; Lanzino, M.; et al. Expression and function of phosphodiesterase type 5 in human breast cancer cell lines and tissues: Implications for targeted therapy. Clin. Cancer Res. 2016, 22, 2271–2282. [Google Scholar] [CrossRef] [Green Version]
- Piazza, G.A.; Thompson, W.J.; Pamukcu, R.; Alila, H.W.; Whitehead, C.M.; Liu, L.; Fetter, J.R.; Gresh, W.E.J.; Klein-Szanto, A.J.; Farnell, D.R.; et al. Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer Res. 2011, 61, 3961–3968. [Google Scholar]
- Pusztai, L.; Zhen, J.H.; Arun, B.; Rivera, E.; Whitehead, C.; Thompson, W.J.; Nealy, K.M.; Gibbs, A.; Symmans, W.F.; Esteva, F.J.; et al. Phase 1 and 2 study of exisulind in combination with capecitabine in patients with metastatic breast cancer. J. Clin. Oncol. 2003, 21, 3454–3461. [Google Scholar] [CrossRef]
- Whitehead, C.M.; Earle, K.A.; Fetter, J.; Xu, S.; Hartman, T.; Chan, D.C.; Zhao, T.L.; Piazza, G.; Klein-Szanto, A.J.; Pamukcu, R.; et al. Exisulind-induced apoptosis in a non-small cell lung cancer orthotopic lung tumor model augments docetaxel treatment and contributes to increased survival. Mol. Cancer Ther. 2003, 2, 479–488. [Google Scholar]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Andò, S.; Catalano, S. Phosphodiesterase Type 5 and Cancers: Progress and Challenges. Oncotarget 2017, 8, 99179–99202. [Google Scholar] [CrossRef] [Green Version]
- Savai, R.; Pullamsetti, S.S.; Banat, G.A.; Weissmann, N.; Ghofrani, H.A.; Grimminger, F.; Schermuly, R.T. Targeting cancer with phosphodiesterase inhibitors. Expert Opin. Investig. Drugs 2010, 19, 117–131. [Google Scholar] [CrossRef]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Catalano, S.; Andò, S. Phosphodiesterase type 5 as a candidate therapeutic target in cancers. Curr. Pathobiol. Rep. 2015, 3, 193–201. [Google Scholar] [CrossRef]
- Goluboff, E.T.; Shabsigh, A.; Saidi, J.A.; Weinstein, I.B.; Mitra, N.; Heitjan, D.; Piazza, G.A.; Pamukcu, R.; Buttyan, R.; Olsson, C.A. Exisulind (sulindac sulfone) suppresses growth of human prostate cancer in a nude mouse xenograft model by increasing apoptosis. Urology 1999, 53, 440–445. [Google Scholar] [CrossRef]
- Baetek, J.; Mistrik, M.; Bartkova, J. Long-distance inflammatory and genotoxic impact of cancer in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 17861–17862. [Google Scholar]
- Das, A.; Durrant, D.; Mitchell, C.; Dent, P.; Batra, S.K.; Kukreja, R.C. Sildenafil (Viagra) sensitizes prostate cancer cells to doxorubicin-mediated apoptosis through CD95. Oncotarget 2016, 7, 4399–4413. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Mei, L.; Fan, X.; Tang, C.; Ji, X.; Hu, X.; Shi, W.; Qian, Y.; Hussain, W.J.; Wang, C.; et al. Phosphodiesterase 5/protein kinase G signal governs stemness of prostate cancer stem cells through Hippo pathway. Cancer Lett. 2016, 378, 38–50. [Google Scholar] [CrossRef]
- Zenzmaier, C.; Sampson, N.; Pernkopf, D.; Plas, E.; Untergasser, G.; Berger, P. Attenuated proliferation and trans-differentiation of prostatic stromal cells indicate suitability of phosphodiesterase type 5 inhibitors for prevention and treatment of benign prostatic hyperplasia. Endocrinology 2010, 151, 3975–3984. [Google Scholar] [CrossRef] [Green Version]
- Chavez, A.H.; ScottCoffield, K.; Hasan Rajab, M.; Jo, C. Incidence rate of prostate cancer in men treated for erectile dysfunction with phosphodiesterase type 5 inhibitors: Retrospective analysis. Asian J. Androl. 2013, 15, 246–248. [Google Scholar] [CrossRef] [Green Version]
- Zelefsky, M.J.; Fuks, M.J.; Hunt, M.; Lee, H.J.; Lombardi, D.; Ling, C.C.; Reuter, V.E.; Venkatraman, E.S.; Leibel, S.A. High dose radiation delivered by intensity modulated conformal radiotherapy improves the outcome of localized prostate cancer. J. Urol. 2001, 166, 876–881. [Google Scholar] [CrossRef]
- Morais-Santos, M.; Werneck-Gomes, H.; Campolina-Silva, G.H.; Santos, L.C.; Mahecha, G.A.B.; Hess, R.A.; Oliveira, C.A. Basal Cells Show Increased Expression of Aromatase and Estrogen Receptor α in Prostate Epithelial Lesions of Male Aging Rats. Endocrinology 2018, 159, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Ellem, S.J.; Risbridger, G.P. Aromatase and prostate cancer. Minerva Endocrinol. 2006, 31, 1–12. [Google Scholar]
- Aversa, A.; Caprio, M.; Antelmi, A.; Armani, A.; Brama, M.; Greco, E.A.; Francomano, D.; Calanchini, M.; Spera, G.; Di Luigi, L.; et al. Exposure to phosphodiesterase type 5 inhibitors stimulates aromatase expression in human adipocytes in vitro. J. Sex. Med. 2011, 8, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Aversa, A.; Fittipaldi, S.; Bimonte, V.M.; Wannenes, F.; Papa, V.; Francomano, D.; Greco, E.A.; Lenzi, A.; Migliaccio, S. Tadalafil modulates aromatase activity and androgen receptor expression in a human osteoblastic cell in vitro model. J. Endocrinol. Investig. 2016, 39, 199–205. [Google Scholar] [CrossRef]
- Aversa, A.; Fittipaldi, S.; Francomano, D.; Bimonte, V.M.; Greco, E.A.; Crescioli, C.; Di Luigi, L.; Lenzi, A.; Migliaccio, S. Tadalafil improves lean mass and endothelial function in normal men with mild ED/LUTS: In vivo and in vitro characterization. Endocrine 2017, 56, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.W.; Johnson, R.T.; Mohler, J.L.; French, F.S.; Wilson, E.M. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001, 61, 2892–2898. [Google Scholar] [PubMed]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen Receptor Gene Expression in Prostate Cancer Is Directly Suppressed by the Androgen Receptor Through Recruitment ofLysine-Specific Demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen Receptor: Structure, Role in Prostate Cancer and Drug Discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Kuruma, H.; Matsumoto, H.; Shiota, M.; Bishop, J.; Lamoureux, F.; Thomas, C.; Briere, D.; Los, G.; Gleave, M.; Fanjul, A.; et al. A novel antiandrogen, Compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Masiello, D.; Cheng, S.; Bubley, G.J.; Lu, M.L.; Balk, S.P. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 2002, 277, 26321–26326. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Stocco, C. Tissue Physiology and Pathology of Aromatase. Steroids 2012, 77, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Cardone, A.; Angelini, F.; Varriale, B. Autoregulation of estrogen and androgen receptor mRNAs and downregulation of androgen receptor mRNA by estrogen in primary cultures of lizard testis cells. Gen. Comp. Endocrinol. 1998, 110, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent global patterns in prostate cancer incidence and mortality rates. Eur. Urol. 2020, 77, 38. [Google Scholar] [CrossRef] [PubMed]
- Peak, T.C.; Yafi, F.A.; Sangkum, P.; Hellstrom, W.J. Emerging drugs for the treatment of erectile dysfunction. Exp. Opin. Emerg. Drugs 2015, 20, 263. [Google Scholar] [CrossRef] [PubMed]
- Kügler, R.; Mietens, A.; Seidensticker, M.; Tasch, S.; Wagenlehner, F.M.; Kaschtanow, A.; Tjahjono, Y.; Tomczyk, C.U.; Beyer, D.; Risbridger, G.P.; et al. Novel imaging of the prostate reveals spontaneous gland contraction and excretory duct quiescence together with different drug effects. FASEB J. 2018, 32, 1130. [Google Scholar] [CrossRef] [Green Version]
- Fibbi, B.; Morelli, A.; Vignozzi, L.; Filippi, S.; Chavalmane, A.; De Vita, G.; Marini, M.; Gacci, M.; Vannelli, G.B.; Sandner, P.; et al. Characterization of phosphodiesterase type 5 expression and functional activity in the human male lower urinary tract. J. Sex. Med. 2010, 7, 59. [Google Scholar] [CrossRef]
- Ückert, S.; Küthe, A.; Jonas, U.D.O.; Stief, C.G. Characterization and functional relevance of cyclic nucleotide phosphodiesterase isoenzymes of the human prostate. J. Urol. 2001, 166, 2484. [Google Scholar] [CrossRef]
- Zhang, W.; Zang, N.; Jiang, Y.; Chen, P.; Wang, X.; Zhang, X. Upregulation of phosphodiesterase type 5 in the hyperplastic prostate. Sci. Rep. 2015, 5, 17888. [Google Scholar] [CrossRef] [Green Version]
- Bisegna, C.; Gravina, G.L.; Pierconti, F.; Martini, M.; Larocca, L.; Rossi, P.; Grimaldi, P.; Di Stasi, S.; Jannini, E.A. Regulation of PDE5 expression in normal prostate, benign prostatic hyperplasia, and adenocarcinoma. Andrology 2020, 8, 427. [Google Scholar] [CrossRef]
- Chang, J.F.; Hsu, J.L.; Sheng, Y.H.; Leu, W.J.; Yu, C.C.; Chan, S.H.; Chan, M.L.; Hsu, L.C.; Liu, S.P.; Guh, J.H. Phosphodiesterase Type 5 (PDE5) Inhibitors sensitize topoisomerase II inhibitors in killing prostate cancer through PDE5-independent impairment of HR and NHEJ DNA repair systems. Front. Oncol. 2019, 8, 681. [Google Scholar] [CrossRef] [Green Version]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [Green Version]
- Sumanasuriya, S.; De Bono, J. Treatment of Advanced Prostate Cancer-A Review of Current Therapies and Future Promise. Cold Spring Harb. Perspect. Med. 2018, 8, a030635. [Google Scholar] [CrossRef]
- Bonkhoff, H.; Berges, R. The evolving role of estrogens and their receptors in the development and progression of prostate cancer. Eur. Urol. 2009, 55, 533. [Google Scholar] [CrossRef]
- Bonkhoff, H. Estrogen receptor signaling in prostate cancer: Implications for carcinogenesis and tumor progression. Prostate 2018, 78, 2. [Google Scholar] [CrossRef]
- Park, K.; Dalton, J.T.; Narayanan, R.; Barbieri, C.E.; Hancock, M.L.; Bostwick, D.G.; Steiner, M.S.; Rubin, M.A. TMPRSS2:ERG gene fusion predicts subsequent detection of prostate cancer in patients with high-grade prostatic intraepithelial neoplasia. J. Clin. Oncol. 2014, 32, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Cao, J.; Tian, L.; Shen, Y.; Yang, X.; Lin, Q.; Zhang, R.; Liu, H.; Du, X.; Shi, J.; et al. Aromatase-induced Endogenous Estrogen Promotes Tumour Metastasis Through Estrogen Receptor-α/Matrix Metalloproteinase 12 Axis Activation in Castration-Resistant Prostate Cancer. Cancer Lett. 2019, 467, 72–84. [Google Scholar] [CrossRef]
- Migliaccio, S.; Newbold, R.R.; Teti, A.; Jefferson, W.J.; Toverud, S.U.; Taranta, A.; Bullock, B.C.; Suggs, C.A.; Spera, G.; Korach, K.S. Transient estrogen exposure of female mice during early development permanently affects osteoclastogenesis in adulthood. Bone 2000, 27, 47–52. [Google Scholar] [CrossRef]
- Krapf, J.M.; Simon, J.A. A sex-specific dose-response curve for testosterone: Could excessive testosterone limit sexual interaction in women? Menopause 2017, 24, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.M.; Kölsch, M.; Larra, M.F.; Zech, C.M.; Blumenthal, T.D.; Frings, C.; Schächinger, H. For whom the bell (curve) tolls: Cortisol rapidly affects memory retrieval by an inverted U-shaped dose-response relationship. Psychoneuroendocrinology 2013, 38, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Greco, W.R.; Bravo, G.; Parsons, J.C. The search for synergy: A critical review from a response surface perspective. Pharmacol. Rev. 1995, 47, 331–385. [Google Scholar]
- Ostano, P.; Mello-Grand, M.; Sesia, D.; Gregnanin, I.; Peraldo-Neia, C.; Guana, F.; Jachetti, E.; Farsetti, A.; Chiorino, G. Gene Expression Signature Predictive of Neuroendocrine Transformation in Prostate Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 1078. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Kaufman, D.; George, D.; Oh, W.K.; Kazanis, M.; Manola, J.; Kantoff, P.W. Selective aromatase inhibition for patients with androgen-independent prostate carcinoma. Cancer 2002, 95, 1864. [Google Scholar] [CrossRef] [PubMed]
- Attia, D.M.A.; Ederveen, A.G.H. Opposing roles of ERα and ERβ in the genesis and progression of adenocarcinoma in the rat ventral prostate. Prostate 2012, 72, 1022. [Google Scholar] [CrossRef] [PubMed]
- Dicitore, A.; Grassi, E.S.; Borghi, M.O.; Gelmini, G.; Cantone, M.C.; Gaudenzi, G.; Persani, L.; Caraglia, M.; Vitale, G. Antitumor activity of interferon-β1a in hormone refractory prostate cancer with neuroendocrine differentiation. J. Endocrinol. Investig. 2017, 40, 761. [Google Scholar] [CrossRef] [PubMed]
- Sadar, M.D. The role of cAMP in regulating the androgen receptor. In Androgen Actions in Prostate Cancer; Tindall, D., Mohler, J., Eds.; Springer Science + Business Media LLC: Heidelberg, Germany, 2009; 465p. [Google Scholar]
- Fu, M.; Wang, C.; Reutens, A.T.; Wang, J.; Angeletti, R.H.; Siconolfi-Baez, L.; Ogryzko, V.; Avantaggiati, M.L.; Pestell, R.G. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J. Biol. Chem. 2000, 275, 20853–20860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Wang, C.; Wang, J.; Zhang, X.; Sakamaki, T.; Yeung, Y.G.; Chang, C.; Hopp, T.; Fuqua, S.A.; Jaffray, E.; et al. Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol. Cell. Biol. 2002, 22, 3373–3388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannattasio, S.; Megiorni, F.; Di Nisio, V.; Del Fattore, A.; Fontanella, R.; Camero, S.; Antinozzi, C.; Festuccia, C.; Gravina, G.L.; Cecconi, S.; et al. Testosterone-mediated activation of androgenic signalling sustains in vitro the transformed and radioresistant phenotype of rhabdomyosarcoma cell lines. J. Endocrinol. Investig. 2019, 42, 183–197. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bimonte, V.M.; Marampon, F.; Antonioni, A.; Fittipaldi, S.; Ferretti, E.; Pestell, R.G.; Curreli, M.; Lenzi, A.; Vitale, G.; Brunetti, A.; et al. Phosphodiesterase Type-5 Inhibitor Tadalafil Modulates Steroid Hormones Signaling in a Prostate Cancer Cell Line. Int. J. Mol. Sci. 2021, 22, 754. https://doi.org/10.3390/ijms22020754
Bimonte VM, Marampon F, Antonioni A, Fittipaldi S, Ferretti E, Pestell RG, Curreli M, Lenzi A, Vitale G, Brunetti A, et al. Phosphodiesterase Type-5 Inhibitor Tadalafil Modulates Steroid Hormones Signaling in a Prostate Cancer Cell Line. International Journal of Molecular Sciences. 2021; 22(2):754. https://doi.org/10.3390/ijms22020754
Chicago/Turabian StyleBimonte, Viviana M., Francesco Marampon, Ambra Antonioni, Simona Fittipaldi, Elisabetta Ferretti, Richard G. Pestell, Mariaignazia Curreli, Andrea Lenzi, Giovanni Vitale, Antonio Brunetti, and et al. 2021. "Phosphodiesterase Type-5 Inhibitor Tadalafil Modulates Steroid Hormones Signaling in a Prostate Cancer Cell Line" International Journal of Molecular Sciences 22, no. 2: 754. https://doi.org/10.3390/ijms22020754