Disulfiram (Antabuse) Activates ROS-Dependent ER Stress and Apoptosis in Oral Cavity Squamous Cell Carcinoma

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Reagents
2.3. Carbonyl Assay
2.4. 35S. labeling and TCA Precipitation
2.5. Proliferation and Cell Death
2.6. Immunoblotting
2.7. RT-qPCR
2.8. Xenografts and Bioluminescent Imaging (BLI)
2.9. Statistical Analysis
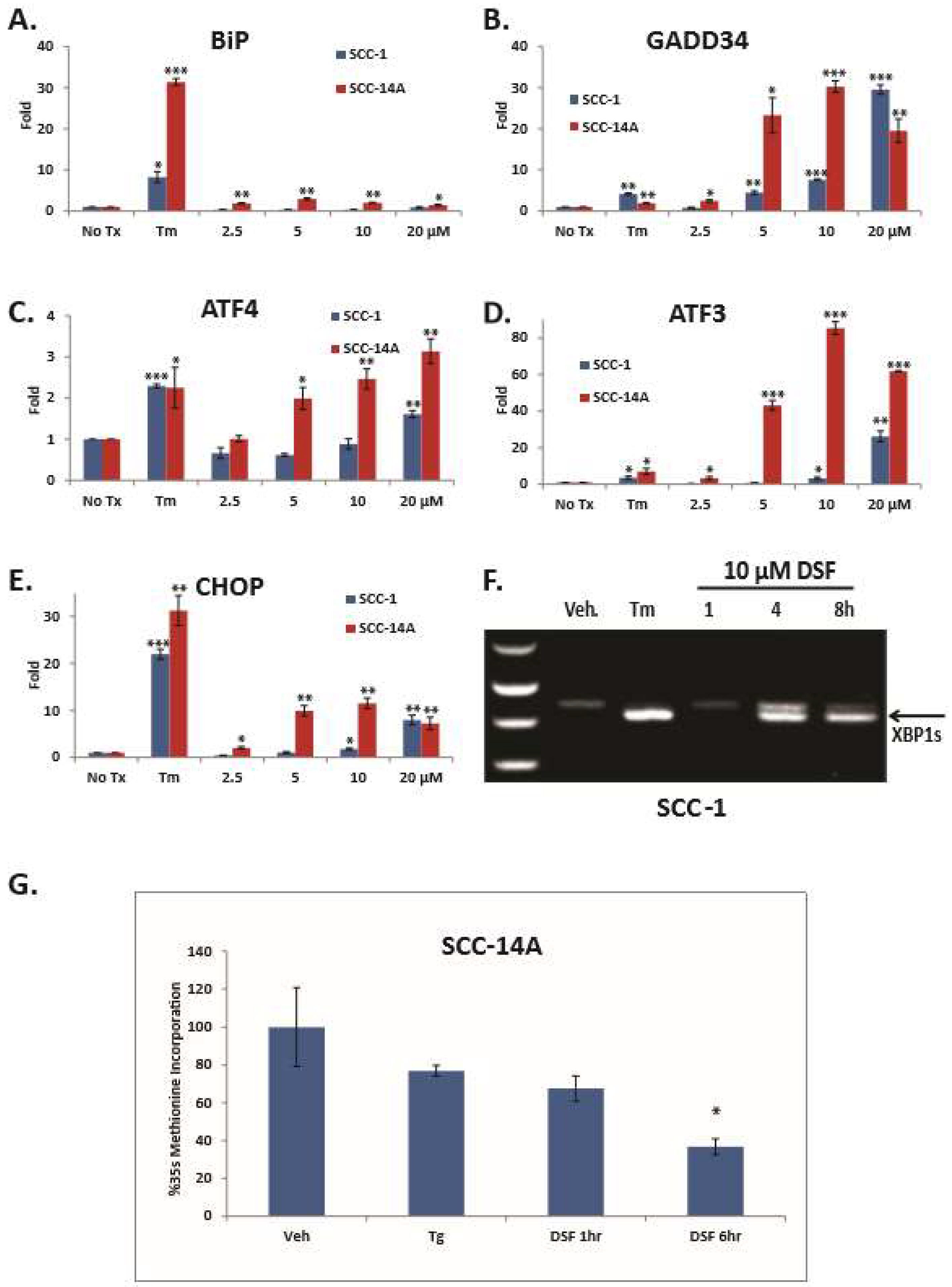
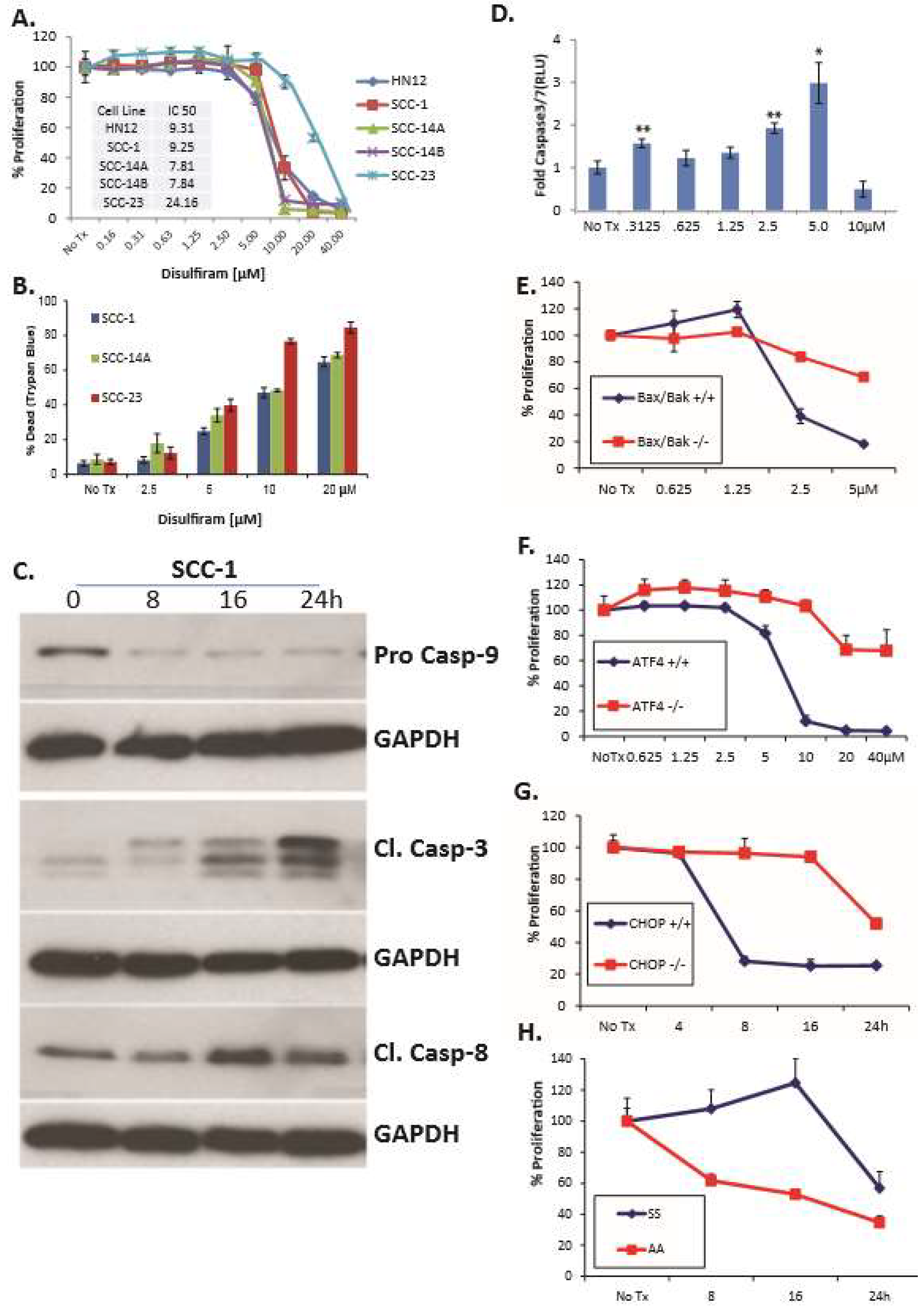
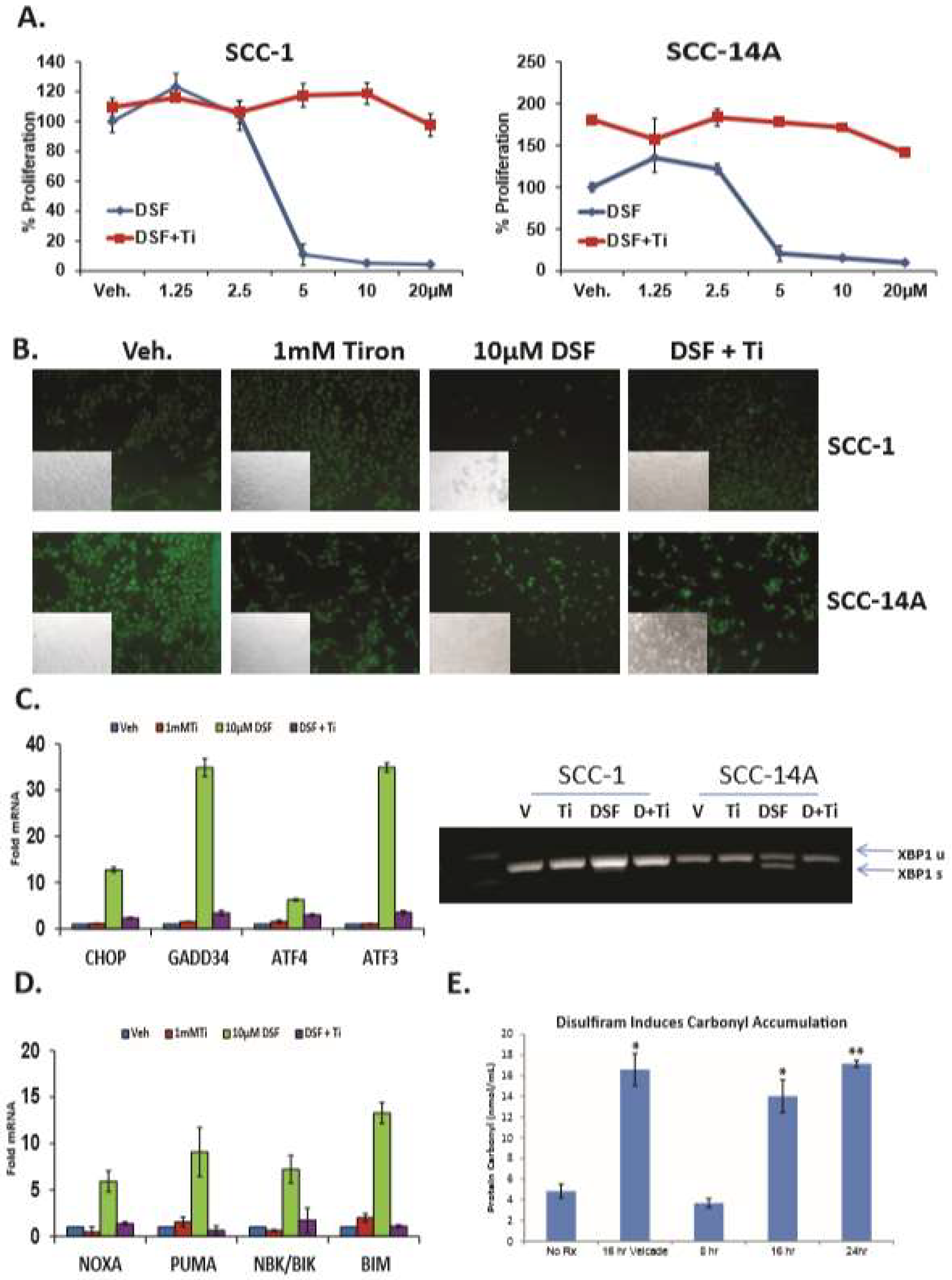
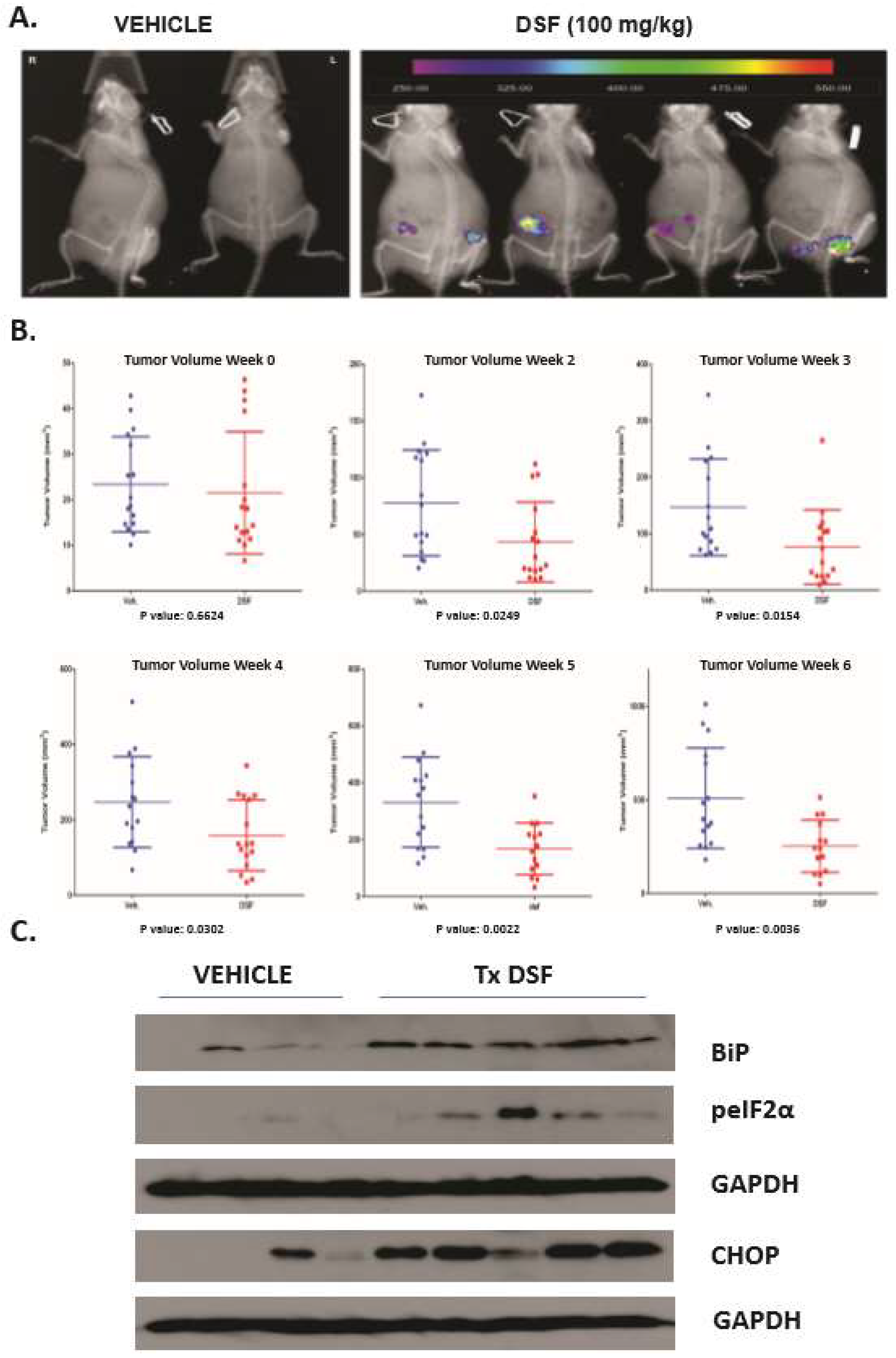
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oral Cancer Foundation. Available online: https://oralcancerfoundation.org/facts/ (accessed on 27 March 2019).
- La Vecchia, C.; Tavani, A.; Franceschi, S.; Levi, F.; Corrao, G.; Negri, E. Epidemiology and prevention of oral cancer. Oral Oncol. 1997, 33, 302–312. [Google Scholar] [CrossRef]
- Wight, A.J.; Ogden, G.R. Possible mechanisms by which alcohol may influence the development of oral cancer--a review. Oral Oncol. 1998, 34, 441–447. [Google Scholar] [CrossRef]
- Ogden, G.R.; Wight, A.J. Aetiology of oral cancer: Alcohol. Br. J. Oral Maxillofac. Surg. 1998, 36, 247–251. [Google Scholar] [CrossRef]
- Hubbers, C.U.; Akgul, B. HPV and cancer of the oral cavity. Virulence 2015, 6, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Nanavati, R.; Modi, T.G.; Dobariya, C. Oral cancer: Etiology and risk factors: A review. J. Cancer Res. Ther. 2016, 12, 458–463. [Google Scholar] [CrossRef]
- Ramsey, T.; Hojjat, H.; Yuhan, B.; Svider, P.F.; Eloy, J.A.; Folbe, A.J.; Raza, S.N.; Fribley, A.M. Disparities in impact of nasopharyngeal cancer: An analysis of global health burden. Laryngoscope 2019. [Google Scholar] [CrossRef]
- Ramsey, T.; Guo, E.; Svider, P.F.; Lin, H.; Syeda, S.; Raza, S.N.; Fribley, A.M. Laryngeal cancer: Global socioeconomic trends in disease burden and smoking habits. Laryngoscope 2018, 128, 2039–2053. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Miller, J.R.; Tripathi, A.; Garshott, D.M.; Brownell, A.L.; Chiego, D.J.; Arevang, C.; Zeng, Q.; Jackson, L.C.; Bechler, S.A.; et al. Borrelidin Induces the Unfolded Protein Response in Oral Cancer Cells and Chop-Dependent Apoptosis. ACS Med. Chem. Lett. 2015, 6, 1122–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, D.P.; Miller, J.R.; Garshott, D.M.; Hedrick, M.; Gosalia, P.; Li, Y.; Milewski, M.; Sugarman, E.; Vasile, S.; Salaniwal, S.; et al. Discovery of Sulfonamidebenzamides as Selective Apoptotic CHOP Pathway Activators of the Unfolded Protein Response. ACS Med. Chem. Lett. 2014, 5, 1278–1283. [Google Scholar] [CrossRef]
- Xi, Y.; Garshott, D.M.; Brownell, A.L.; Yoo, G.H.; Lin, H.S.; Freeburg, T.L.; Yoo, N.G.; Kaufman, R.J.; Callaghan, M.U.; Fribley, A.M. Cantharidins induce ER stress and a terminal unfolded protein response in OSCC. J. Dent. Res. 2015, 94, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Topisirovic, I.; Guzman, M.L.; McConnell, M.J.; Licht, J.D.; Culjkovic, B.; Neering, S.J.; Jordan, C.T.; Borden, K.L. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol. Cell Biol. 2003, 23, 8992–9002. [Google Scholar] [CrossRef]
- Assouline, S.; Culjkovic, B.; Cocolakis, E.; Rousseau, C.; Beslu, N.; Amri, A.; Caplan, S.; Leber, B.; Roy, D.C.; Miller, W.H., Jr.; et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): A proof-of-principle clinical trial with ribavirin. Blood 2009, 114, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, I.B.; Chen, J.J.; Wang, S.; Savas, L.; London, I.M.; Pullman, J. Upregulation of protein synthesis initiation factor eIF-4E is an early event during colon carcinogenesis. Oncogene 1999, 18, 2507–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerekatte, V.; Smiley, K.; Hu, B.; Smith, A.; Gelder, F.; De Benedetti, A. The proto-oncogene/translation factor eIF4E: A survey of its expression in breast carcinomas. Int. J. Cancer 1995, 64, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Franklin, S.; Pho, T.; Abreo, F.W.; Nassar, R.; De Benedetti, A.; Stucker, F.J.; Nathan, C.O. Detection of the proto-oncogene eIF4E in larynx and hypopharynx cancers. Arch. Otolaryngol. Head Neck Surg. 1999, 125, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.O.; Liu, L.; Li, B.D.; Abreo, F.W.; Nandy, I.; De Benedetti, A. Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene 1997, 15, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Karagöz, G.E.; Fröhlich, F.; Li, H.; Walther, T.C.; Walter, P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife 2018, 7, e43036. [Google Scholar] [CrossRef]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fribley, A.M.; Cruz, P.G.; Miller, J.R.; Callaghan, M.U.; Cai, P.; Narula, N.; Neubig, R.R.; Showalter, H.D.; Larsen, S.D.; Kirchhoff, P.D.; et al. Complementary cell-based high-throughput screens identify novel modulators of the unfolded protein response. J. Biomol. Screen 2011, 16, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta. Psychiatr. Scand. Suppl. 1992, 369, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.P.; Mays, D.C.; Lipsky, J.J. Inhibition of recombinant human mitochondrial and cytosolic aldehyde dehydrogenases by two candidates for the active metabolites of disulfiram. Biochemistry 1997, 36, 13748–13754. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.G.; Engel, C.; Wheatley, C.; Nielsen, J. Modification of the sensitivity and repair of potentially lethal damage by diethyldithiocarbamate during and following exposure of plateau-phase cultures of mammalian cells to radiation and cis-diamminedichloroplatinum(II). Cancer Res. 1982, 42, 3074–3078. [Google Scholar]
- Hacker, M.P.; Ershler, W.B.; Newman, R.A.; Gamelli, R.L. Effect of disulfiram (tetraethylthiuram disulfide) amd diethyldithiocarbamate on the bladder toxicity and antitumor activity of cyclophosphamide in mice. Cancer Res. 1982, 42, 4490–4494. [Google Scholar] [PubMed]
- Bodenner, D.L.; Dedon, P.C.; Keng, P.C.; Katz, J.C.; Borch, R.F. Selective protection against cis-diamminedichloroplatinum(II)-induced toxicity in kidney, gut, and bone marrow by diethyldithiocarbamate. Cancer Res. 1986, 46, 2751–2755. [Google Scholar]
- Yip, N.C.; Fombon, I.S.; Liu, P.; Brown, S.; Kannappan, V.; Armesilla, A.L.; Xu, B.; Cassidy, J.; Darling, J.L.; Wang, W. Disulfiram modulated ROS-MAPK and NFkappaB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br. J. Cancer 2011, 104, 1564–1574. [Google Scholar] [CrossRef]
- Fribley, A.; Zeng, Q.; Wang, C.Y. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol. Cell Biol. 2004, 24, 9695–9704. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Fribley, A.M.; Miller, J.R.; Reist, T.E.; Callaghan, M.U.; Kaufman, R.J. Large-scale analysis of UPR-mediated apoptosis in human cells. Methods Enzymol. 2011, 491, 57–71. [Google Scholar] [Green Version]
- Fribley, A.; Zhang, K.; Kaufman, R.J. Regulation of apoptosis by the unfolded protein response. Methods Mol. Biol. 2009, 559, 191–204. [Google Scholar] [PubMed]
- Park, J.W.; Woo, K.J.; Lee, J.T.; Lim, J.H.; Lee, T.J.; Kim, S.H.; Choi, Y.H.; Kwon, T.K. Resveratrol induces pro-apoptotic endoplasmic reticulum stress in human colon cancer cells. Oncol. Rep. 2007, 18, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Bassanetti, I.; Bignardi, C.; Elviri, L.; Tegoni, M.; Mucchino, C.; Bussolati, O.; Franchi-Gazzola, R.; Marchiò, L. Copper binding agents acting as copper ionophores lead to caspase inhibition and paraptotic cell death in human cancer cells. J. Am. Chem. Soc. 2011, 133, 6235–6242. [Google Scholar] [CrossRef] [PubMed]
- Hulan, H.W.; Proudfoot, F.G. Effects of light source, ambient temperature, and dietary energy source on the general performance and incidence of leg abnormalities of roaster chickens. Poult. Sci. 1987, 66, 645–651. [Google Scholar] [CrossRef]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Oliver, C.N. Metal-catalyzed oxidation of proteins. Physiological consequences. J. Biol. Chem. 1991, 266, 2005–2008. [Google Scholar] [PubMed]
- Williams, D.E. Effects of alcohol on workers with carbon disulfide. JAMA 1937, 109, 2. [Google Scholar]
- Hald, J.A.J.E. A drug sensitizing the organism to ethyl alcohol. Lancet 1948, 25, 4. [Google Scholar]
- Martensen-Larsen, O.; Copenhagen, M.D. Treatment of alcoholism with a sensitising drug. Lancet 1948, 252, 1004–1005. [Google Scholar] [CrossRef]
- Paranjpe, A.; Zhang, R.; Ali-Osman, F.; Bobustuc, G.C.; Srivenugopal, K.S. Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis 2014, 35, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Triscott, J.; Lee, C.; Hu, K.; Fotovati, A.; Berns, R.; Pambid, M.; Luk, M.; Kast, R.E.; Kong, E.; Toyota, E. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget 2012, 3, 1112–1123. [Google Scholar] [CrossRef]
- Chiba, T.; Suzuki, E.; Yuki, K.; Zen, Y.; Oshima, M.; Miyagi, S.; Saraya, A.; Koide, S.; Motoyama, T.; Ogasawara, S.; et al. Disulfiram eradicates tumor-initiating hepatocellular carcinoma cells in ROS-p38 MAPK pathway-dependent and -independent manners. PLoS ONE 2014, 9, e84807. [Google Scholar] [CrossRef]
- Huang, H.; Liao, Y.; Liu, N.; Hua, X.; Cai, J.; Yang, C.; Long, H.; Zhao, C.; Chen, X.; Lan, X.; et al. Two clinical drugs deubiquitinase inhibitor auranofin and aldehyde dehydrogenase inhibitor disulfiram trigger synergistic anti-tumor effects in vitro and in vivo. Oncotarget 2016, 7, 2796–2808. [Google Scholar] [CrossRef]
- Brar, S.S.; Grigg, C.; Wilson, K.S.; Holder, W.D., Jr.; Dreau, D.; Austin, C.; Foster, M.; Ghio, A.J.; Whorton, A.R.; Stowell, G.W.; et al. Disulfiram inhibits activating transcription factor/cyclic AMP-responsive element binding protein and human melanoma growth in a metal-dependent manner in vitro, in mice and in a patient with metastatic disease. Mol. Cancer Ther. 2004, 3, 1049–1060. [Google Scholar]
- Lin, J.; Haffner, M.C.; Zhang, Y.; Lee, B.H.; Brennen, W.N.; Britton, J.; Kachhap, S.K.; Shim, J.S.; Liu, J.O.; Nelson, W.G.; et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate 2011, 71, 333–343. [Google Scholar] [CrossRef]
- Han, J.; Liu, L.; Yue, X.; Chang, J.; Shi, W.; Hua, Y. A binuclear complex constituted by diethyldithiocarbamate and copper(I) functions as a proteasome activity inhibitor in pancreatic cancer cultures and xenografts. Toxicol. Appl. Pharmacol. 2013, 273, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Shi, P.; Fombon, I.S.; Zhang, Y.; Huang, F.; Wang, W.; Zhou, S. Disulfiram/copper complex activated JNK/c-jun pathway and sensitized cytotoxicity of doxorubicin in doxorubicin resistant leukemia HL60 cells. Blood Cells Mol. Dis. 2011, 47, 264–269. [Google Scholar] [CrossRef]
- Xu, B.; Wang, S.; Li, R.; Chen, K.; He, L.; Deng, M.; Kannappan, V.; Zha, J.; Dong, H.; Wang, W. Disulfiram/copper selectively eradicates AML leukemia stem cells in vitro and in vivo by simultaneous induction of ROS-JNK and inhibition of NF-kappaB and Nrf2. Cell Death Dis. 2017, 8, e2797. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cui, Q.C.; Yang, H.; Dou, Q.P. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Z.; Brown, S.; Kannappan, V.; Tawari, P.E.; Jiang, W.; Irache, J.M.; Tang, J.Z.; Armesilla, A.L.; Darling, J.L. Liposome encapsulated Disulfiram inhibits NFkappaB pathway and targets breast cancer stem cells in vitro and in vivo. Oncotarget 2014, 5, 7471–7485. [Google Scholar] [CrossRef] [PubMed]
- Cheriyan, V.T.; Wang, Y.; Muthu, M.; Jamal, S.; Chen, D.; Yang, H.; Polin, L.A.; Tarca, A.L.; Pass, H.I.; Dou, Q.P. Disulfiram suppresses growth of the malignant pleural mesothelioma cells in part by inducing apoptosis. PLoS ONE 2014, 9, e93711. [Google Scholar] [CrossRef]
- Jivan, R.; Damelin, L.H.; Birkhead, M.; Rousseau, A.L.; Veale, R.B.; Mavri-Damelin, D. Disulfiram/copper-disulfiram Damages Multiple Protein Degradation and Turnover Pathways and Cytotoxicity is Enhanced by Metformin in Oesophageal Squamous Cell Carcinoma Cell Lines. J. Cell Biochem. 2015, 116, 2334–2343. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clay, M.R.; Tabor, M.; Owen, J.H.; Carey, T.E.; Bradford, C.R.; Wolf, G.T.; Wicha, M.S.; Prince, M.E. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck 2010, 32, 1195–1201. [Google Scholar] [CrossRef]
- Joshua, B.; Kaplan, M.J.; Doweck, I.; Pai, R.; Weissman, I.L.; Prince, M.E.; Ailles, L.E. Frequency of cells expressing CD44, a head and neck cancer stem cell marker: Correlation with tumor aggressiveness. Head Neck 2012, 34, 42–49. [Google Scholar] [CrossRef]
- Curtarelli, R.B.; Gonçalves, J.M.; Dos Santos, L.G.P.; Savi, M.G.; Nör, J.E.; Mezzomo, L.A.M.; Rodríguez Cordeiro, M.M. Expression of Cancer Stem Cell Biomarkers in Human Head and Neck Carcinomas: A Systematic Review. Stem Cell Rev. 2018, 14, 769–784. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, Y.W.; Hsu, H.S.; Tseng, L.M.; Huang, P.I.; Lu, K.H.; Chen, D.T.; Tai, L.K.; Yung, M.C.; Chang, S.C.; et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res. Commun. 2009, 385, 307–313. [Google Scholar] [CrossRef]
- Kulsum, S.; Sudheendra, H.V.; Pandian, R.; Ravindra, D.R.; Siddappa, G.; R, N.; Chevour, P.; Ramachandran, B.; Sagar, M.; Jayaprakash, A.; et al. Cancer stem cell mediated acquired chemoresistance in head and neck cancer can be abrogated by aldehyde dehydrogenase 1 A1 inhibition. Mol. Carcinog. 2017, 56, 694–711. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G.; Palaniyandi, S.; Clark, C.; Nathan, C.O.; Abreo, F.W.; Caldito, G. Analysis of eIF4E and 4EBP1 mRNAs in head and neck cancer. Laryngoscope 2011, 121, 2136–2141. [Google Scholar] [CrossRef]
- Garshott, D.M.; Bechler, S.A.; Burchhardt, D.M.; Shah Obrien, P.; Yoo, G.H.; Chiego, D.J.; Rehman, A.O.; Callaghan, M.U.; Fribley, A.M. The Unfolded Protein Response as a Therapeutic Target for Head and Neck Squamous Cell Carcinoma. In Targeting Oral Cancer; Fribley, A.M., Ed.; Springer International Publishing: Berlin, Germany, 2016; pp. 225–261. [Google Scholar]
- Dudek, J.; Benedix, J.; Cappel, S.; Greiner, M.; Jalal, C.; Müller, L.; Zimmermann, R. Functions and pathologies of BiP and its interaction partners. Cell Mol. Life Sci. 2009, 66, 1556–1569. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef]
- Lopez-Lazaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox. Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah O’Brien, P.; Xi, Y.; Miller, J.R.; Brownell, A.L.; Zeng, Q.; Yoo, G.H.; Garshott, D.M.; O’Brien, M.B.; Galinato, A.E.; Cai, P.; et al. Disulfiram (Antabuse) Activates ROS-Dependent ER Stress and Apoptosis in Oral Cavity Squamous Cell Carcinoma. J. Clin. Med. 2019, 8, 611. https://doi.org/10.3390/jcm8050611
Shah O’Brien P, Xi Y, Miller JR, Brownell AL, Zeng Q, Yoo GH, Garshott DM, O’Brien MB, Galinato AE, Cai P, et al. Disulfiram (Antabuse) Activates ROS-Dependent ER Stress and Apoptosis in Oral Cavity Squamous Cell Carcinoma. Journal of Clinical Medicine. 2019; 8(5):611. https://doi.org/10.3390/jcm8050611
Chicago/Turabian StyleShah O’Brien, Priyanka, Yue Xi, Justin R. Miller, Amy L. Brownell, Qinghua Zeng, George H. Yoo, Danielle M. Garshott, Matthew B. O’Brien, Anthony E. Galinato, Peter Cai, and et al. 2019. "Disulfiram (Antabuse) Activates ROS-Dependent ER Stress and Apoptosis in Oral Cavity Squamous Cell Carcinoma" Journal of Clinical Medicine 8, no. 5: 611. https://doi.org/10.3390/jcm8050611