 Tomáš Takáč
Tomáš Takáč Jozef Šamaj
Jozef Šamaj- Department of Cell Biology, Faculty of Science, Centre of the Region Haná for Biotechnological and Agricultural Research, Palacký University, Olomouc, Czech Republic
Introduction
The molecular mechanism of signal transduction by mitogen-activated protein kinases (MAPKs) in plants is an intensive research field of contemporary plant biology. Signaling in plants involves perception of changing environment and the intracellular transduction of such generated information leading to the activation of adaptive mechanisms or programmed cell death, as it is in the case of hypersensitive response to some biotrophic pathogen attack. At the molecular level, distinct signaling pathways, such as MAPK modules, involve reversible phosphorylation, enzymatic activation, protein-protein interaction and finally transcriptional activation via nuclear-resident transcription factors or cell reorganization via regulation of cytoplasmic or plasma membrane-associated proteins. All these processes are strictly regulated and subcellularly compartmentalized.
Modern OMICS technologies encompassing transcriptomic (Krysan et al., 2002; Frei dit Frey et al., 2014), metabolomic (Lassowskat et al., 2014), proteomic (Miles et al., 2009; Conroy et al., 2013; Lassowskat et al., 2014; Takáč et al., 2014) and phosphoproteomic (Hoehenwarter et al., 2013; Lassowskat et al., 2014) approaches have been employed to gain functional information on plant signaling. The main challenges of advanced (phospho)proteomic studies are: reliable identification of all signaling constituents of a particular MAPK pathway as well as identification and validation of respective target proteins. In this opinion article, we aim to briefly evaluate benefits and pitfalls of various proteomic approaches that are used to study MAPK signaling in plants. We emphasize on the advantages of comparative shot-gun proteomic analyses of Arabidopsis mutants and transgenic plants with altered MAPK signaling. The importance of critical data validation and interpretation is also discussed.
Proteomic Approaches on MAPK Signaling
Modern proteomics offers a variety of approaches to study cell signaling events in living organisms (Jørgensen and Locard-Paulet, 2012). They have been quite rarely used also for investigation of molecular mechanisms in plant MAPK signaling (Table 1). Nevertheless, these studies substantially helped to better describe complex processes accompanied with signaling and to find new kinase targets regulated by reversible phosphorylation.
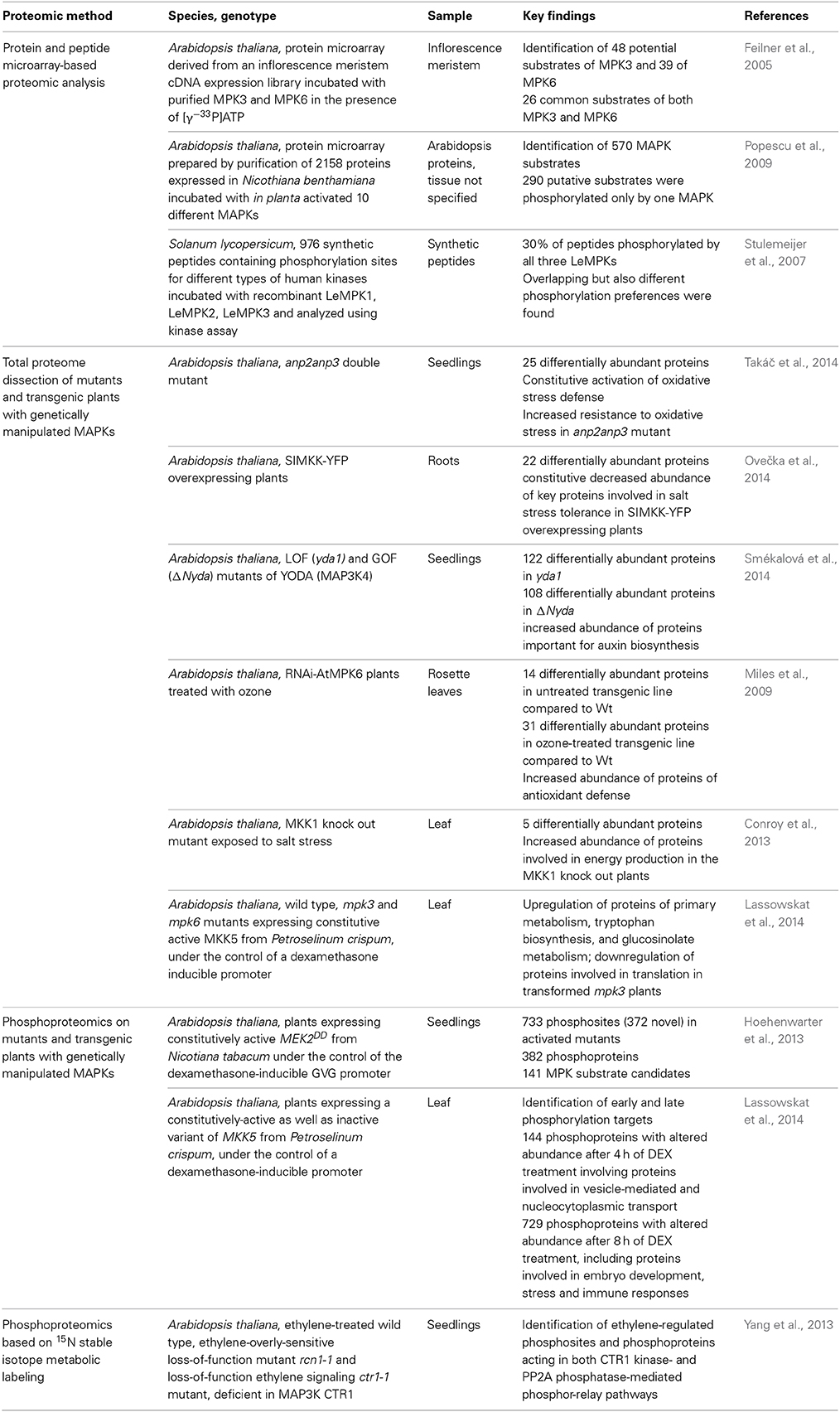
Table 1. Overview of proteomic approaches on MAPK signaling in plants.
Targeted phosphoproteomics is a powerful tool for the identification of putative downstream substrates of MAPKs, but also for mapping of phosphorylation sites detected upon activation or inactivation of particular MAPK pathways (de la Fuente van Bentem et al., 2008; Hoehenwarter et al., 2013; Lassowskat et al., 2014). Diverse phosphoproteomic approaches have been applied, mostly employing genetically modified kinase expression systems. For example, a recent phosphoproteomic study of transgenic Arabidopsis thaliana plants harboring a gene encoding a constitutively active MEKDD from Nicotiana tabacum (NtMEK2DD) expressed under the control of the dexamethasone (DEX)-inducible GVG promoter identified 141 putative MAPK substrates with the help of complementary enrichment of phosphoproteins and consecutive phosphopeptide enrichment (Hoehenwarter et al., 2013). In a similar approach, early and late putative substrates of MPK3 and MPK6 were identified by phosphoproteomics performed on Arabidopsis thaliana plants transformed with a constitutively-active variant of MKK5 from Petroselinum crispum, expressed under the control of a DEX-inducible promoter (Lassowskat et al., 2014). The expression of heterologous MAP2Ks in Arabidopsis might result in the identification of somehow unspecific or indirect targets. This problem might be solved by validation of putative MAPK substrates by both bioinformatic and independent analytical methods.
The phosphoproteomics following stable isotope labeling in Arabidopsis (SILIA) efficiently elucidated a protein complex containing ethylene receptor ETR1 and constitutive triple response 1 (CTR1) MAP3K (Yang et al., 2013). Using the loss of function ethylene response Arabidopsis mutant ctr1-1, novel CTR1 targets involved in ethylene response were identified including calcium-sensing receptor and plastidal transcriptionally active protein.
Previously, microarray containing 1690 proteins from Arabidopsis was generated and incubated with MAPKs in the presence of [γ−33P]ATP. Quantitative evaluation of radioactive signals identified 48 potential substrates of MPK3 and 39 substrates of MPK6 (Feilner et al., 2005). A more extended protein microarray-based approach for MAPK phosphorylation target analysis was published by Popescu and coworkers (Popescu et al., 2009). First, purified wild type MAPKs (MPK1–MPK8, MPK10, and MPK16) were tested by in vitro phosphorylation assays for their activation by certain MAPKKs co-expressed in Nicothiana benthamiana leaves. Subsequently, 2156 proteins were placed on protein microarray and probed by above MAPKs. This approach allowed the identification of new signaling modules composed of MAPKKs, MAPKs and their target proteins.
Peptide microarray analysis based on screening of hundreds of synthetic peptides containing experimentally verified phosphorylation sites for different types of human kinases was used to discriminate between Solanum lycopersicum LeMPK1, LeMPK2, and LeMPK3 (Stulemeijer et al., 2007). The incubation of peptide microarrays with recombinant LeMPK1, LeMPK2, and LeMPK3 in the presence of [γ−33P]ATP was followed by in vitro kinase assay which displayed both similar, but also different peptides phosphorylated by these MAPKs.
In our opinion, another promising tool for MAPK substrate identification was provided by recent study, which used a library of 377 synthetic peptides, representing previously identified phosphorylation sites in developing seeds of Arabidopsis thaliana and Brassica napus, for screening with purified kinases while the resulting phospho-sites were analyzed subsequently by mass spectrometry (Ahsan et al., 2013). It is of great value that all these diverse phosphoproteomic approaches helped to identify new putative targets of MAPKs. However, in our view they lack the capability of shot-gun comparative proteomics to identify mid- and long-term complex protein profiles and networks associated with the transcriptional impact of deregulation of MAPK pathways, as this seems to be often the case in the mutant and overexpressor plant lines (e.g., Krysan et al., 2002). We consider this point quite important because changed complex protein profiles revealed by shot-gun proteomics might be closely linked to phenotypes of respective mutant and transgenic lines, including their characteristics such as modified stress responses or developmental defects.
Benefits and Weaknesses of Comparative Shot-Gun Proteomics on Arabidopsis MAPK Mutants and Transgenic Overexpression Plants
Complex molecular processes affected by deregulation of MAPK signaling cascades in planta are often focused on transcriptomic studies (Frei dit Frey et al., 2014) while global protein changes are still underestimated. Since conditionally-modulated transcript levels can be rectified at the protein level, it is of essential importance to perform proteomics rather than speculate on protein abundance based on transcriptomic data (Gygi et al., 1999). Shot-gun proteomic analyses on mutant and transgenic plants with genetically modified MAPKs and their upstream activators such as MAP2Ks and MAP3Ks provide a very useful proteome readout and quite unique information because they might not be fully consistent with transcriptomic data from the same plant lines (e.g., Krysan et al., 2002 cf. Takáč et al., 2014). Few studies used shot-gun proteomic approaches to better explain stress-related and developmental phenotypes in Arabidopsis mutants and transgenic lines with altered MAPKs expression. One earlier study employing ICAT (isotope-coded affinity tag) quantitative proteomic analysis on MPK6 RNAi transgenic Arabidopsis plants unveiled molecular mechanisms accompanying response to ozone fumigation in relation to MPK6 downregulation (Miles et al., 2009). The redox response pathway was different between untreated plants with suppressed MPK6 expression and the corresponding control wild type plants. After ozone treatment, a more pronounced antioxidant defense was observed in plants with downregulated MPK6 (Miles et al., 2009). Recently, the two-dimensional LC MS/MS analysis of Arabidopsis double mutant deficient in two redundant MAP3Ks called ANP2 and ANP3 revealed constitutive upregulation of a protein functional network involved in oxidative stress response in the double mutant anp2anp3 plants (Takáč et al., 2014). This suggested an increased resistance of the anp2anp3 mutant against oxidative stress which was corroborated by showing increased viability of this mutant growing on paraquat-supplemented medium. The activation of a protein network consisting of superoxide dismutase isoforms, chaperonin 20 and enzymes of the ascorbate-glutathione cycle was validated through detailed biochemical, physiological and histochemical analyses showing connection of proteomic data with resilience of the above mutant to oxidative stress. Importantly, this shot-gun proteomic study uncovered a previously unknown function of ANP2 and ANP3 in Arabidopsis antioxidant defense.
Next, we reported that the heterologous overexpression of alfalfa SIMKK-YFP leads to salt hypersensitivity of Arabidopsis seedlings. The proteomic analysis showed decreased levels of several key proteins involved in salt tolerance in the roots of these SIMKK-overexpressor transgenic plants (Ovečka et al., 2014). Thus, the overexpression of SIMKK repressed the levels of catalase, peroxiredoxin, glutathione S-transferase, nucleoside diphosphate kinase 1, endoplasmic reticulum luminal-binding protein 2, and finally those of plasma membrane aquaporins. Downregulation of these proteins most likely contributed to the increased salt sensitivity of transgenic Arabidopsis seedlings overexpressing SIMKK (Ovečka et al., 2014).
In addition to the analysis of stress responses, shot-gun proteomic analyses on mutant and transgenic plants might help to explain unique growth or developmental phenotypes regulated by members of particular MAPK pathway. Recently, we have found that both inactivation and constitutive activation of MAP3K called YODA caused pronounced root phenotypes in Arabidopsis (Smékalová et al., 2014). Analysis of endogenous auxin levels showed that these phenotypes might be related to increased IAA production in respective yda1 and ΔNyda mutants. Again, shot-gun proteomics provided insights to the molecular mechanism underlying auxin overproduction by detection of elevated levels of proteins involved in auxin biosynthesis such as tryptophan synthase in ΔNyda and nitrilases in both mutants (Smékalová et al., 2014).
Finally, a recent elegant study used integrated metabolomics, shot-gun proteomics and phosphoproteomics approaches on leaves of Arabidopsis Col-0 ecotype as well as mpk3 and mpk6 mutants, all expressing DEX-inducible constitutively active Petroselinum crispum MKK5 (MKK5DD). Heterologous MKK5DD expression activated MPK3 and MPK6 at equal levels in Col-0 plants. The induction of MKK5DD in the mpk6 mutant resulted in more robust proteome changes compared to mpk3. In this case, shot-gun proteomic data were in positive correlation with metabolomic as well as phosphoproteomic analyses (Lassowskat et al., 2014).
All these shot-gun proteomic studies helped to identify new molecular processes linked to MAPK pathways. This capability could be assigned to phosphoproteomics as well, as it was shown for example for nitrogen metabolism, control of circadian clock and phototropism (Hoehenwarter et al., 2013), and for biosynthesis of antimicrobial defense substances (Lassowskat et al., 2014).
Proteomics on total protein extract has technically limited capability for identification of all molecular mechanisms connected with particular MAPK pathway. These limitations arise from limited proteome resolution especially in terms of detection of low abundant proteins and inability of this approach to resolve phosphorylation events. Nevertheless, we consider these studies feasible in order to elucidate molecular networks composed from abundant proteins, including those involved in antioxidant defense (Miles et al., 2009; Takáč et al., 2014) or stress response (Ovečka et al., 2014). In our view shot-gun proteomics can also provide important initial insights into new biological processes regulated by MAPKs, which should be later investigated by targeted application of genetic and cell biological methods. In ideal case, general shot-gun differential proteomics may be perhaps quite efficiently complemented by targeted phosphoproteomic (Lassowskat et al., 2014). In this respect, dual-enrichment techniques (Hoehenwarter et al., 2013; Lassowskat et al., 2014) might represent a crucial tool in order to overcome the limitation of the large dynamic range of protein abundance in shot-gun proteomic approaches.
Evaluation and Validation of Proteomic Data
The complexity of proteomic analysis and the high inter-replicate variability of the proteomic data necessitate proper independent validation methods to confirm proteomic results. This significantly increases the biological value of the proteomic analysis. For example, protein abundance might be verified using immunoblotting with monospecific primary antibodies (Takáč et al., 2014). As an alternative, immunolocalization of protein in intact respective tissue have been also employed (Takáč et al., 2013) and provide useful information about spatial protein localization. The live cell microscopy imaging using fluorescently tagged proteins may be also used to study protein localization (Takáč et al., 2012). Nevertheless, to study abundance of fluorescently tagged protein of interest it should be expressed under the control of its own promoter in the respective mutant background (Smékalová et al., 2014). Transgenic mutant lines with such construct have to show rescue of the mutant phenotype which is considered as a proof of construct functionality. Subsequently, such rescued mutants bearing tagged protein can be used for isolation of functional protein complexes and MS analysis. To validate enzymatic activity, specific spectrophotometric measurements and/or activity staining on native polyacrylamide gels might be used (Takáč et al., 2014). The validation of protein phosphorylation is possible via kinase assays (Feilner et al., 2005; Yang et al., 2013; Lassowskat et al., 2014), by using Phos-tag technology (Kinoshita et al., 2006; Komis et al., 2014) or by isoelectric focusing combined with immunoblotting (Anderson and Peck, 2008). Protein-protein interactions can be tested by BIFC or co-immunoprecipitation assays (Singh et al., 2012).
The efficiency of shot-gun proteomics for the exploration of MAPK signaling pathways could be significantly increased by bioinformatic evaluation of proteomic data. There are several commercial software and web-based applications providing various analyses including functional categorization of differentially abundant proteins (Conesa and Götz, 2008), clustering of commonly expressed proteins (de Hoon et al., 2004), metabolic pathway analysis tools (Iersel et al., 2008), applications summarizing protein-protein interactions (Jensen et al., 2009), coexpressing protein candidates (Obayashi and Kinoshita, 2010), tools for prediction of protein localization (Horton et al., 2007), detection of signaling peptides (Petersen et al., 2011), search for most abundant sequence motifs (Chou and Schwartz, 2011) and others.
Goal-Oriented Approaches and Complementarity in Complex Cell Signaling Analysis
As explained above, the high throughput analyses of MAPK signaling such as transcriptomics, proteomics and phosphoproteomics are not always necessarily converging to a common output. They have, at least partially, different scopes and their results might be either complementary or contradictory (Gygi et al., 1999). Protein array surveys identify a list of putative substrates of MAPKs but outside of a physiological context. In this sense, phosphoproteomic analyses will decipher phosphoproteins in MAPK mutants or under conditions triggering MAPK activity but with uncertainty as to whether they truly constitute exclusive MAPK substrates (they might be eventually phosphorylated also by alternative kinases). Finally, proteomics (including shot-gun approaches) has potential to corroborate the transcriptional transactivation role of MAPK signaling and should provide factual data on the protein levels. Although proteomics will not reveal mechanistic insights of MAPK signaling (i.e., identify MAPK substrates) it will provide the landscape of its physiological consequences which can be efficiently followed by targeted biochemical, genetic cell biological and physiological assays.
Altogether, this article summarizes recent evidence that shot-gun proteomic analysis of Arabidopsis mutants and transgenic lines with genetically-manipulated MAPK expression is a reliable method for the detection of some molecular networks accompanying developmental or conditional phenotypes of these mutants and lines. We strongly recommend using proper independent methods, including biochemical, cell biological and phosphoproteomic ones, for validation and corroboration of proteomic data. Such detailed studies may eventually provide new links between MAPK-related changes in protein abundance and direct regulation of cellular processes through MAPK-dependent phosphorylation of target proteins.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Program for Sustainability I (NPU grant no. LO1204) to the Centre of the Region Haná for Biotechnological and Agricultural Research. We would like to thank George Komis for useful suggestions and critical reading of the manuscript.
References
Ahsan, N., Huang, Y., Tovar-Mendez, A., Swatek, K. N., Zhang, J., Miernyk, J. A., et al. (2013). A Versatile mass spectrometry-based method to both identify kinase client-relationships and characterize signaling network topology. J. Proteome Res. 12, 937–948. doi: 10.1021/pr3009995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anderson, J. C., and Peck, S. C. (2008). A simple and rapid technique for detecting protein phosphorylation using one-dimensional isoelectric focusing gels and immunoblot analysis. Plant J. 55, 881–885. doi: 10.1111/j.1365-313X.2008.03550.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chou, M. F., and Schwartz, D. (2011). Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinformatics Chapter 13, Unit 13. 15–24. doi: 10.1002/0471250953.bi1315s35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conesa, A., and Götz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832. doi: 10.1155/2008/619832
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conroy, C., Ching, J., Gao, Y., Wang, X., Rampitsch, C., and Xing, T. (2013). Knockout of AtMKK1 enhances salt tolerance and modifies metabolic activities in Arabidopsis. Plant Signal. Behav. 8:e24206. doi: 10.4161/psb.24206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Hoon, M. J. L., Imoto, S., Nolan, J., and Miyano, S. (2004). Open source clustering software. Bioinformatics 20, 1453–1454. doi: 10.1093/bioinformatics/bth078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de la Fuente van Bentem, S., Mentzen, W. I., de la Fuente, A., and Hirt, H. (2008). Towards functional phosphoproteomics by mapping differential phosphorylation events in signaling networks. Proteomics 8, 4453–4465. doi: 10.1002/pmic.200800175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feilner, T., Hultschig, C., Lee, J., Meyer, S., Immink, R. G. H., Koenig, A., et al. (2005). High throughput identification of potential Arabidopsis mitogen-activated protein kinases substrates. Mol. Cell. Proteomics 4, 1558–1568. doi: 10.1074/mcp.M500007-MCP200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frei dit Frey, N., Garcia, A. V., Bigeard, J., Zaag, R., Bueso, E., Garmier, M., et al. (2014). Functional analysis of Arabidopsis immune-related MAPKs uncovers a role for MPK3 as negative regulator of inducible defences. Genome Biol. 15:R87. doi: 10.1186/gb-2014-15-6-r87
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gygi, S. P., Rochon, Y., Franza, B. R., and Aebersold, R. (1999). Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730.
Hoehenwarter, W., Thomas, M., Nukarinen, E., Egelhofer, V., Röhrig, H., Weckwerth, W., et al. (2013). Identification of novel in vivo MAP kinase substrates in Arabidopsis thaliana through use of tandem metal oxide affinity chromatography. Mol. Cell. Proteomics 12, 369–380. doi: 10.1074/mcp.M112.020560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horton, P., Park, K.-J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iersel, M. P., van Kelder, T., Pico, A. R., Hanspers, K., Coort, S., Conklin, B. R., et al. (2008). Presenting and exploring biological pathways with PathVisio. BMC Bioinformatics 9:399. doi: 10.1186/1471-2105-9-399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jensen, L. J., Kuhn, M., Stark, M., Chaffron, S., Creevey, C., Muller, J., et al. (2009). STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416. doi: 10.1093/nar/gkn760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jørgensen, C., and Locard-Paulet, M. (2012). Analysing signalling networks by mass spectrometry. Amino Acids 43, 1061–1074. doi: 10.1007/s00726-012-1293-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kinoshita, E., Kinoshita-Kikuta, E., Takiyama, K., and Koike, T. (2006). Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757. doi: 10.1074/mcp.T500024-MCP200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Komis, G., Takáč, T., Bekešová, S., Vadovič, P., and Samaj, J. (2014). Affinity-Based SDS PAGE identification of phosphorylated Arabidopsis MAPKs and substrates by acrylamide pendant Phos-TAG™. Methods Mol. Biol. 1171, 47–63. doi: 10.1007/978-1-4939-0922-3_5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krysan, P. J., Jester, P. J., Gottwald, J. R., and Sussman, M. R. (2002). An Arabidopsis mitogen-activated protein kinase kinase kinase gene family encodes essential positive regulators of cytokinesis. Plant Cell 14, 1109–1120. doi: 10.1105/tpc.001164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lassowskat, I., Böttcher, C., Eschen-Lippold, L., Scheel, D., and Lee, J. (2014). Sustained mitogen-activated protein kinase activation reprograms defense metabolism and phosphoprotein profile in Arabidopsis thaliana. Front. Plant Sci. 5:554. doi: 10.3389/fpls.2014.00554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miles, G. P., Samuel, M. A., Ranish, J. A., Donohoe, S. M., Sperrazzo, G. M., and Ellis, B. E. (2009). Quantitative proteomics identifies oxidant-induced, AtMPK6-dependent changes in Arabidopsis thaliana protein profiles. Plant Signal. Behav. 4, 497–505. doi: 10.4161/psb.4.6.8538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Obayashi, T., and Kinoshita, K. (2010). Coexpression landscape in ATTED-II: usage of gene list and gene network for various types of pathways. J. Plant Res. 123, 311–319. doi: 10.1007/s10265-010-0333-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ovečka, M., Takáč, T., Komis, G., Vadovič, P., Bekešová, S., Doskočilová, A., et al. (2014). Salt-induced subcellular kinase relocation and seedling susceptibility caused by overexpression of Medicago SIMKK in Arabidopsis. J. Exp. Bot. 65, 2335–2350. doi: 10.1093/jxb/eru115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Popescu, S. C., Popescu, G. V., Bachan, S., Zhang, Z., Gerstein, M., Snyder, M., et al. (2009). MAPK target networks in Arabidopsis thaliana revealed using functional protein microarrays. Genes Dev. 23, 80–92. doi: 10.1101/gad.1740009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, R., Lee, M.-O., Lee, J.-E., Choi, J., Park, J. H., Kim, E. H., et al. (2012). Rice mitogen-activated protein kinase interactome analysis using the yeast two-hybrid system. Plant Physiol. 160, 477–487. doi: 10.1104/pp.112.200071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smékalová, V., Luptovčiak, I., Komis, G., Šamajová, O., Ovečka, M., Doskočilová, A., et al. (2014). Involvement of YODA and mitogen activated protein kinase 6 in Arabidopsis post-embryogenic root development through auxin up-regulation and cell division plane orientation. New Phytol. 203, 1175–1193. doi: 10.1111/nph.12880
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stulemeijer, I. J. E., Stratmann, J. W., and Joosten, M. H. A. J. (2007). Tomato mitogen-activated protein kinases LeMPK1, LeMPK2, and LeMPK3 are activated during the cf-4/avr4-induced hypersensitive response and have distinct phosphorylation specificities. Plant Physiol. 144, 1481–1494. doi: 10.1104/pp.107.101063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takáč, T., Pechan, T., Šamajová, O., and Šamaj, J. (2013). Vesicular trafficking and stress response coupled to PI3K inhibition by LY294002 as revealed by proteomic and cell biological analysis. J. Proteome Res. 12, 4435–4448. doi: 10.1021/pr400466x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takáč, T., Pechan, T., Samajova, O., Ovecka, M., Richter, H., Eck, C., et al. (2012). Wortmannin treatment induces changes in Arabidopsis root proteome and post-golgi compartments. J. Proteome Res. 11, 3127–3142. doi: 10.1021/pr201111n
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takáč, T., Šamajová, O., Vadovič, P., Pechan, T., Košútová, P., Ovečka, M., et al. (2014). Proteomic and biochemical analyses show functional network of proteins involved in antioxidant defense of Arabidopsis anp2anp3 double mutant. J. Proteome Res. 13, 5347–5361. doi: 10.1021/pr500588c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Z., Guo, G., Zhang, M., Liu, C. Y., Hu, Q., Lam, H., et al. (2013). Stable isotope metabolic labeling-based quantitative phosphoproteomic analysis of Arabidopsis mutants reveals ethylene-regulated time-dependent phosphoproteins putative substrates of constitutive triple response 1 kinase. Mol. Cell. Proteomics 12, 3559–3582. doi: 10.1074/mcp.M113.031633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mitogen-activated protein kinase (MAPK), signaling, proteomics, phosphoproteomics, shot-gun proteomics, validation
Citation: Takáč T and Šamaj J (2015) Advantages and limitations of shot-gun proteomic analyses on Arabidopsis plants with altered MAPK signaling. Front. Plant Sci. 6:107. doi: 10.3389/fpls.2015.00107
Received: 18 December 2014; Accepted: 09 February 2015;
Published online: 25 February 2015.
Edited by:
Joshua L. Heazlewood, The University of Melbourne, AustraliaReviewed by:
Justin Lee, Leibniz Institute of Plant Biochemistry, GermanyGerold J. M. Beckers, RWTH Aachen University, Germany
Copyright © 2015 Takáč and Šamaj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: jozef.samaj@upol.cz